
文章阅读目录大纲
访问在线服务: http://metdna.zhulab.cn/
Metabolite identification is the long-standing challenge for liquid chromatography-mass spectrometry (LC-MS)-based untargeted metabolomics. Here, we developed MetDNA (Metabolite identification and Dysregulated Network Analysis) for the large-scale and ambiguous identification of metabolites from LC-MS/MS data sets. Users can simply import MS1 peak table, MS/MS data and sample information to perform metabolite identification and dysregulated metabolic pathway analysis. MetDNA implements a metabolic reaction network (MRN) based recursive algorithm for metabolite identification, which supports data from different LC systems (e.g., HILIC and reverse phase) and MS platforms (e.g., Agilent QTOF, Sciex TripleTOF, Thermo Orbitrap, and others).
X. Shen, R. Wang, X. Xiong, Y. Yin, Y. Cai, Z. Ma, N. Liu, and Z.-J. Zhu* (Corresponding Author), Metabolic Reaction Network-based Recursive Metabolite Annotation for Untargeted Metabolomics, Nature Communications, 2019, 10: 1516.
按照目前的代谢组学发展程度,在整个数据分析流程之中,对我们的代谢组学数据分析结果制约程度最大的瓶颈存在于将质谱原始数据之中的未知的峰特征进行注释。我们只有在得到对应的代谢物数据库信息之后,从而才能够进行下游的代谢物分类统计,代谢通路分析等与生物学功能相关联的数据分析之中。
目前在代谢组学数据分析之中进行代谢物的注释方法无非就是两种:将样本中得到的二级谱图数据与标准品库之中的参考谱图数据进行比较,综合取得分最好的结果完成注释;另外一个方法则是基于化学信息学分析来完成不依赖参考库的代谢物注释方法。对于这两种代谢物注释方法各有利有弊:
- 对于标准品库数据比对注释方法,基于cos相似度计算的方法非常的经典,得到的具有高分的二级注释结果一般都非常准确。但是目前本方法也会受到标准品费用高昂,并且也并非所有的代谢物都存在有对应的化学合成方法或者纯化方法来获取标准品。所以目前进行依赖标准品库数据的代谢物注释也仅仅能够对1千到两千个代谢物产生注释信息。并且我们进行标准品库注释,一般会限定于某种特定的设备仪器平台上完成,因为质谱数据在不同的仪器平台之间的差异有点大。
- 因为上面所提到的种种缺陷,所以目前在标准品库注释方面的限制很大。在目前科学界内大家一般是将方法研究精力集中在了不依赖于标准品库的注释方法开发上。不依赖标准品库注释,顾名思义就是不依赖于标准品数据做注释,而是根据谱图的聚类性质以及生物学上下文信息来完成对应的谱图信息推导。对于这个方法,虽然注释的准确度比不上基于标准品的注释结果,但是存在有可以不限制于特定的仪器平台,成本低廉的优势。
MetaDNA注释算法
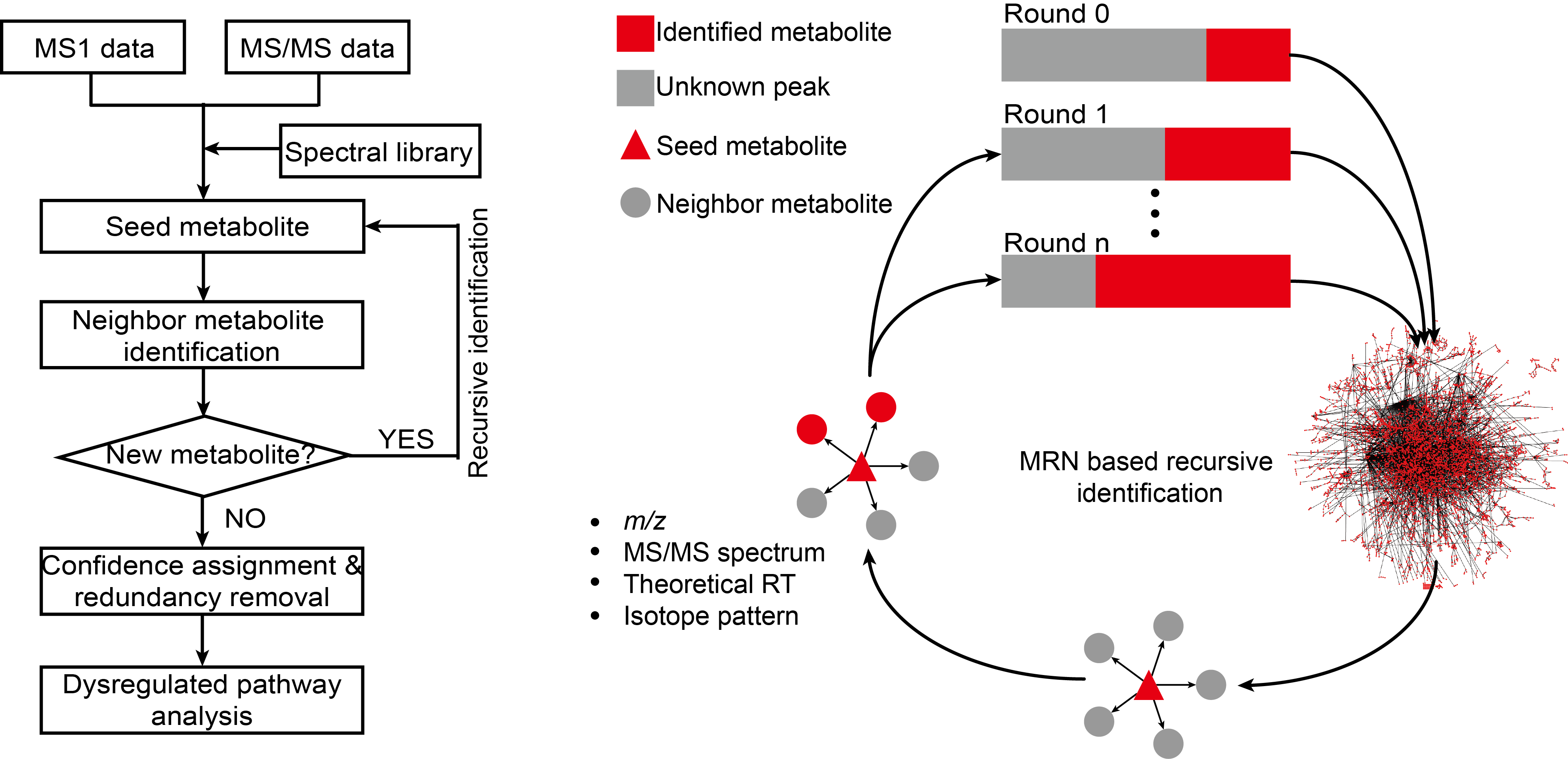
中国科学院上海有机化学研究所生物与化学交叉研究中心朱正江研究员课题组于2019年发表了一个基于KEGG代谢网络的递归式外延二级谱图注释的MetaDNA方法(metabolic reaction network (MRN)-based recursive algorithm (MetDNA))。可以有效地基于生物学上下文信息对样本之中的二级谱图数据进行推导注释。下面我们就来详细的了解一下这个方法的具体实现原理。
首先,我们需要明确的一点是:在MetaDNA算法之中,其非常巧妙的将生物学共表达理论结合了目前在代谢组学质谱数据处理方法之中比较流行的分子网络理论。
生物学共表达理论
一般来说,在细胞内产生的相对应的生物化学过程的单元就是一个生化反应。对于细胞内的生物化学反应过程,一般都是可逆的,所以我们一般都可以认可在化学方程式的左右两端的代谢物都会存在于细胞内,相互各自为对方的反应底物和合成产物。在更高一层次,如果细胞要实现某种生物学功能的话,至少某一个代谢途径之中的化学反应过程是相互联通的。即这些在活跃的代谢途径之中的生化反应都会相互关联的激活在细胞环境之中。所以基于这个生物学假设前提,产生对应的生物学功能的实现,某一个代谢途径中的代谢反应过程相关的代谢物都可能会出现在细胞环境之中。这样子我们做诸如KEGG代谢途径富集,就会对产生某种生物学功能变化的代谢途径产生对应的富集计算结果。
那讲解到这里,大家就会想到了,生物学共表达理论的应用最广泛最成熟的分析方法就是代谢通路富集了。没错了,如果我们不针对差异代谢物做富集分析,而是针对所有注释得到的代谢物做富集分析,实际上就是对我们的代谢物列表做生物学共表达检验计算。
目前基于生物学共表达理论进行代谢物注释的方法中,比较经典的一个方法就是MetaboAnalyst程序包之中所使用到的Mummichog

Li S, Park Y, Duraisingham S, Strobel FH, Khan N, Soltow QA, et al. (2013) Predicting Network Activity from High Throughput Metabolomics. PLoS Comput Biol 9(7): e1003123. https://doi.org/10.1371/journal.pcbi.1003123
基于生物学共表达理论,如果我们的代谢物注释结果是正确的话,则相应的代谢通路富集结果应该是非常显著的。所以在Mummichog

在这个方法之中,采用了一个很简单的富集得分计算方法来计算代谢途径生物共表达:使用属于某一个子图的candidate列表数量除以该子图的代谢物节点数量,再乘上整个网络结果的模块度值(关于网络模块度值的计算方法,大家可以阅读《基于Louvain算法的网络集群发现》了解算法细节)用来描述富集情况。很显然,我们的代谢物注释结果candidate列表越接近子图的代谢物节点数量,并且对应的模块度越高,则整个公式计算得到的富集得分越高,这说明注释得到的代谢物列表可能越准确。
分子网络理论
在质谱计算解谱分析方法中,目前在学术界存在有一种很流行的分析方法:分子网络(Molecular Networking)。分子网络的建立过程非常的简单,我们只需要对拿到的一个大小为N个二级质谱图的集合中的二级谱图数据进行两两cos相似度计算,得到一个大小为N*N
在应用分子网络进行质谱数据分析,以GNPS数据库的建立方法最为经典。在GNPS数据库之中,首先针对一些代谢物进行了标准品的种子数据的构建。然后在这些种子标准品数据的基础之上,不断地将其他的样本谱图数据放入其中进行分子网络的建立。在GNPS数据库之中,二级谱图得到的一簇聚类往往都是化学分类相似的一簇代谢物的结果。所以我们可以在通过GNPS数据库建立的分子网络基础上,可以通过节点与节点间的质量差及中性丢失分析,在GNPS已经鉴定出来的代谢物的基础上进行未知化合物结构的推测。

Aron, A. T., Gentry, E. C., McPhail, K. L., Nothias, L.-F., Nothias-Esposito, M., Bouslimani, A., … Dorrestein, P. C. (2020). Reproducible molecular networking of untargeted mass spectrometry data using GNPS. Nature Protocols. doi:10.1038/s41596-020-0317-5
从上面针对GNPS的数据库构建流程图之中我们也可以了解得到一个现象:化学结构相似的代谢物往往会很可能产生比较相似的质谱碎片信息。所以基于分子网络理论,我们就可以依照质谱图相似度所建立的网络,针对一系列聚类在一块的质谱图进行化学结构注释。
MetaDNA算法讲解
那在了解完了上面的两个基础知识点之后,其实我们就已经将MetaDNA的最基础的计算原理给吃透了。在细胞代谢中,一个代谢物可以通过酶催化反应转变为另一种代谢物。同一个代谢反应中两个代谢物可定义为反应对邻近代谢物(reaction-paired neighbor metabolite)。反应对邻近代谢物具有结构相似性,因此其MS/MS谱图也具有一定的相似性。在MetaDNA方法之中,作者提出了在基元反应过程的左右两端的代谢物的化学结构是相似的,这样子根据分子网络的理论,他们二者产生的二级质谱图理论上也是会比较相似的这样子的一个化学信息学假设,并在对应的METLIN数据库工作之中给出了标准品数据验证。
那既然A到B这样子的一个化学反应过程可以通过两端的代谢物的质谱数据所建立的分子网络匹配上,同样的,我们也可以将B到C这样子的一个化学反应过程按照同样的理论相匹配上。这样子,理论上我们就可以将某一个代谢通路之中的代谢网络通过分子网络所描述出来。
利用这个原理,MetDNA算法利用样本中已经鉴定出的代谢物作为种子,进一步鉴定代谢网络中邻近的代谢物。新鉴定出的代谢物可作为新的种子,继续鉴定代谢网络中邻近代谢物,递归运算,直到不再能够鉴定出新的邻近代谢物。该算法的最大特点是可以通过代谢反应网络去鉴定没有标准MS/MS谱图的代谢物,使得代谢物的结构鉴定并不依赖很大规模的标准MS/MS数据库。

通过前面的一个知识点讲解,我们可以基于生物学共表达理论了解到,一个通路之中的代谢物是会倾向于共同表达来实现对应的代谢物在代谢网络中的流动,从而产生对应的生物学功能。所以在MetaDNA方法之中,就可以依照共表达的代谢物列表,以及分子网络的建立,不断的在整个代谢网络之中进行推断注释过程的传递,从而将样本数据中可能分布在整个代谢网络之中的未知代谢物通过网络上下游的方法,一层一层的都鉴定出来。




- 单细胞视角下的微生物基因组代谢酶嵌入分析 - 2026年2月25日
- 基因组代谢酶层级嵌入 - 2026年2月23日
- 基因组功能吉布斯LDA主题建模 - 2026年2月23日

No responses yet