
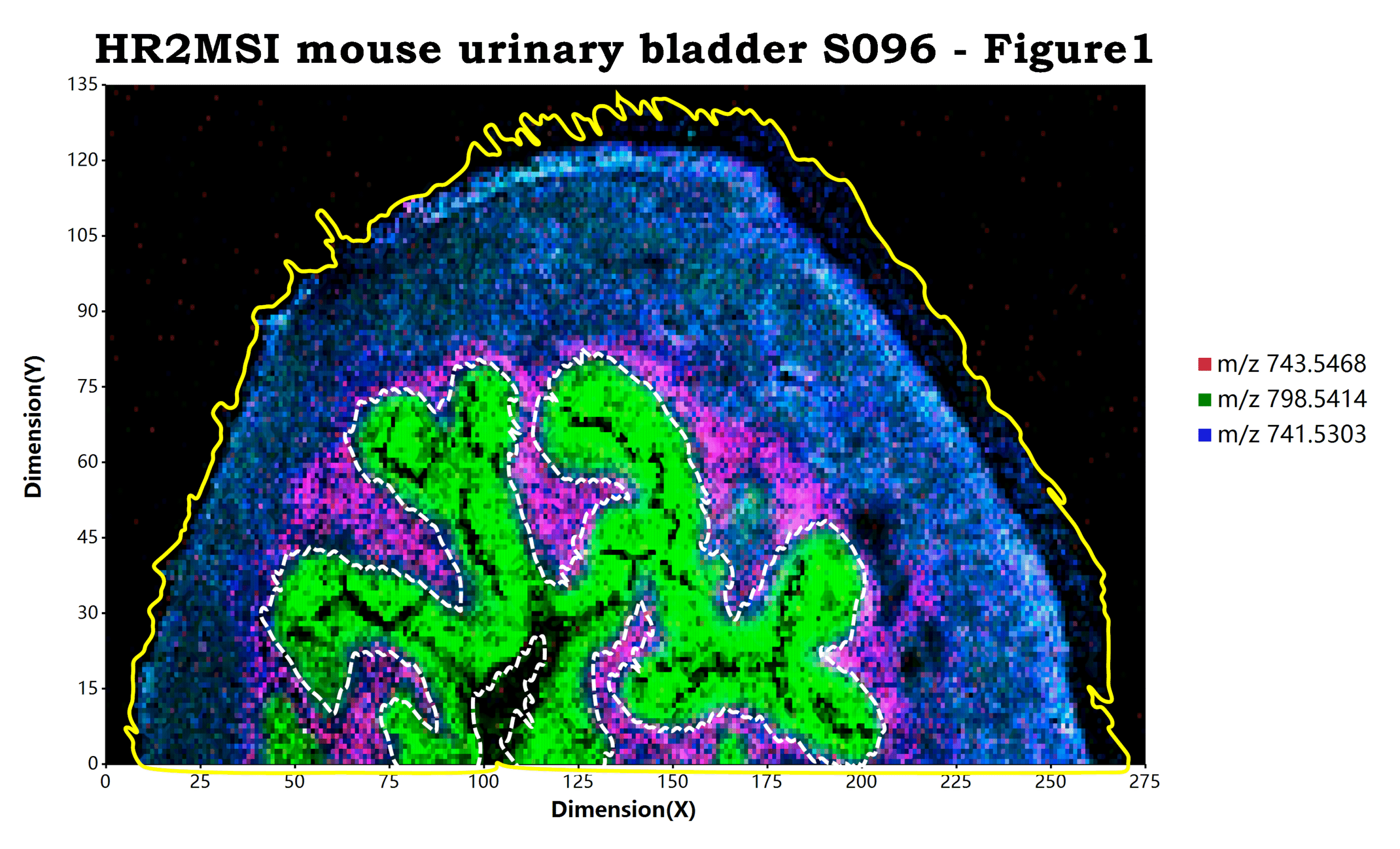
估计阅读时长: 10 分钟目前经过改进和优化之后的基于mzkit代码库底层的msimaging质谱成像软件包在样本可视化上进行了非常多的改进,诸如: 添加样本原始背景叠加 目前进行质谱成像可视化,程序包不仅仅可以使用任意rgb纯色来作为可视化的背景。目前还可以支持直接使用原始数据的背景作为质谱成像的显示背景。进行这个显示的秘诀就在于简单的在脚本中添加一个TIC背景图层:geom_MSIbackground("TIC") ggplot(msi_data, padding = "padding: 200px 600px 200px 250px;") + geom_MSIbackground("TIC") # rendering of […]
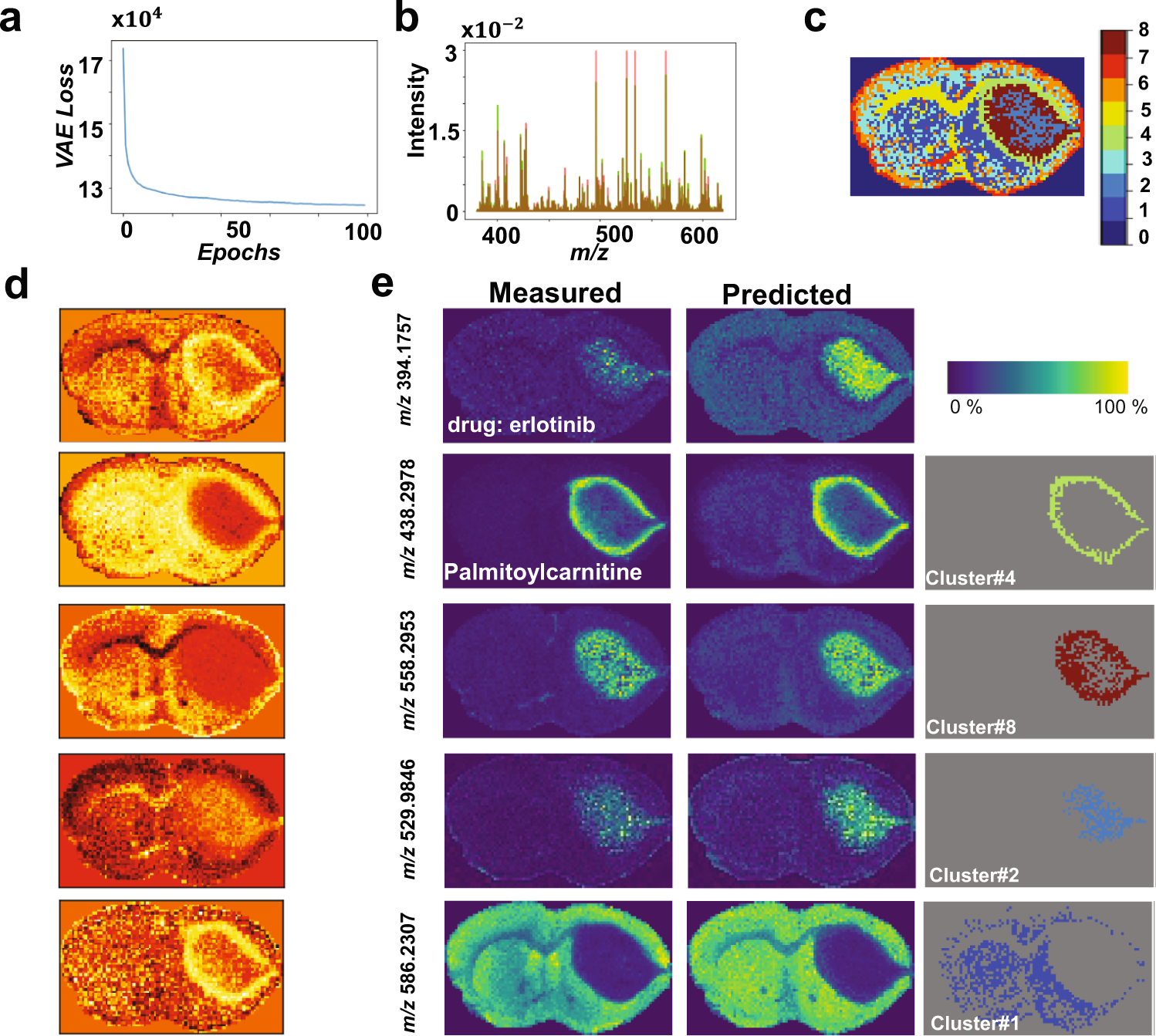
估计阅读时长: 4 分钟基于UMAP工具进行简单的自动化组织分区操作 在这里我们假设已经可以正常的将空间代谢数据导入至MZKit工作站软件之中。假若需要借助于MZKit工作站软件进行切片组织样本的自动化分区操作,相关的功能可以在【MSI Analysis】菜单栏中寻找到。在这里我们打开【Show Map Layer】按钮,选择【UMAP and clustering】功能。 基于降维的组织自动化分区原理 因为降维操作一般是一种特征提取操作,所以经过降维之后,在高维度空间上无法显现的特征,在低维度会呈现出来。在高维度空间散落的相近的数据点,在经过特征提取之后,低维度上会产生相似的特征信息,相互聚集在一簇。这样子我们就可以在低维度空间上通过一些聚类算法讲这些特征进行聚类,最后将聚类特征结果标记到各个散点上的对应的原始成像空间上,我们就可以看见组织分区的结果了。 Abdelmoula, W.M., Lopez, B.GC., Randall, E.C. et […]
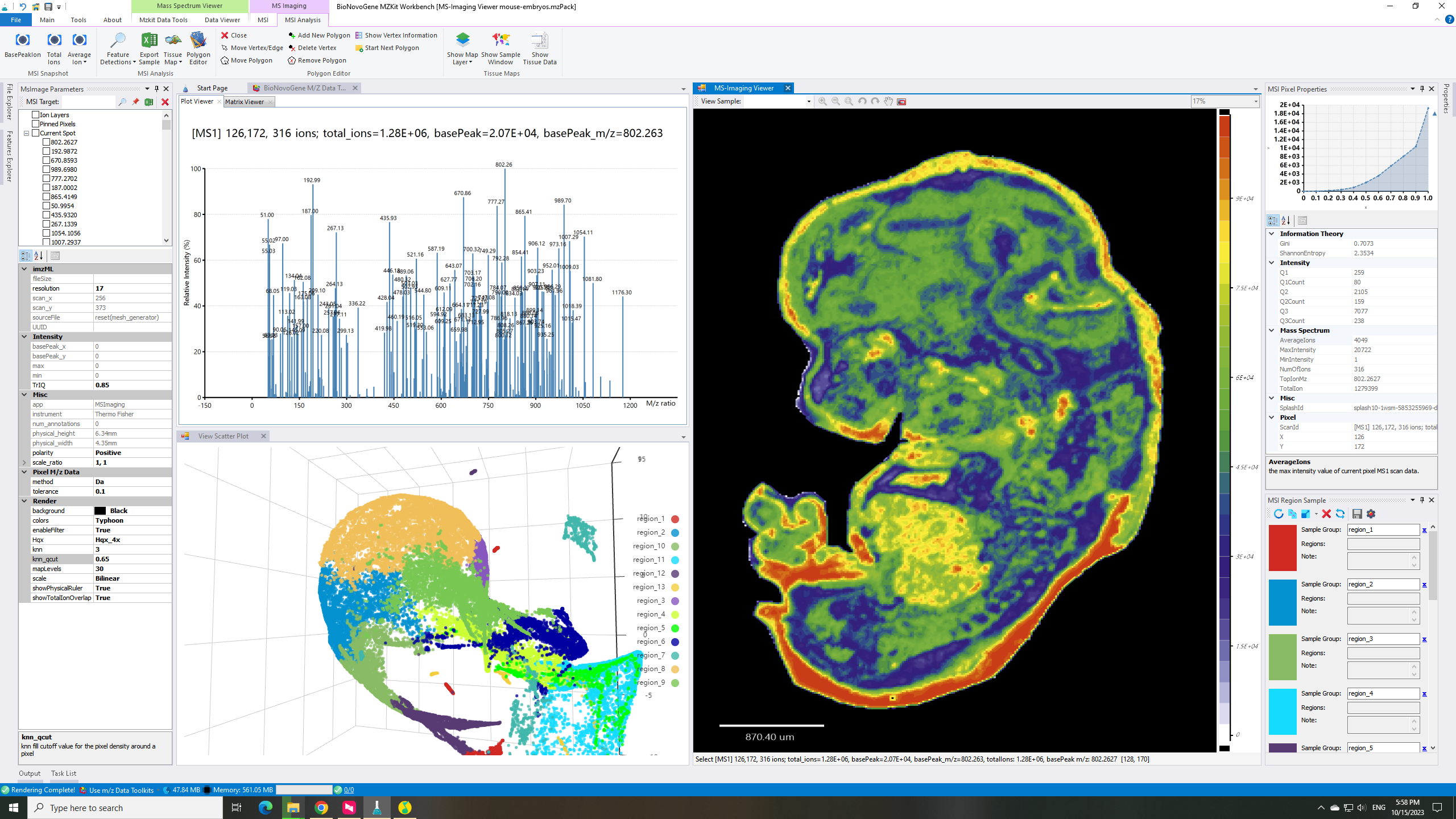
估计阅读时长: 6 分钟大家好呀,今天的这篇文章主要是为了回答在B站上的一位小伙伴的请求 Order by Date Name Attachments render-parameters • 18 kB • 808 click 2023年10月15日view-umap • 427 […]
估计阅读时长: 5 分钟在BILIBILI上观看视频:《【BioNovoGene Mzkit教程】代谢组学原始数据处理基础》 最近我在B站的视频页面下发现了这样的一条评论,面对质谱数据分析领域内的初学者的求教,其实自己也是非常的诚惶诚恐的。因为在视频中所使用的脚本语言是自己开发的一门新语言,所以可能给一些初学者造成了一部分的困扰哈哈😅😄😅😅。首先先对这个粉丝说一声抱歉哈。 针对上述的提问,我的回答大概是有以下的几点: Order by Date Name Attachments question_20230223 • 17 kB • 777 click […]
估计阅读时长: 4 分钟在代谢组学领域内,LCMS原始数据分析一般分为非靶向全扫原始数据,以及仅针对某些离子进行扫描的MRM靶向质谱数据。虽然二者都是基于LCMS方法进行实验,但是MRM靶向数据由于在事先已经通过实验确定,得到了Q1和Q3离子对信息,所以可以仅针对某一些特定代谢物进行检测。因为MRM数据是针对于某些代谢物检测的靶向数据,所以其XIC谱图在没有同分异构体存在的情况下,一般是很纯净的目标化合物的检测结果数据。所以在原始数据分离,定量计算方面都要比非靶向全扫结果数据要容易很多。 Order by Date Name Attachments xcms-logo-white • 183 kB • 841 click 2022年7月1日lcmspreproc_slides_1.2 • 136 […]
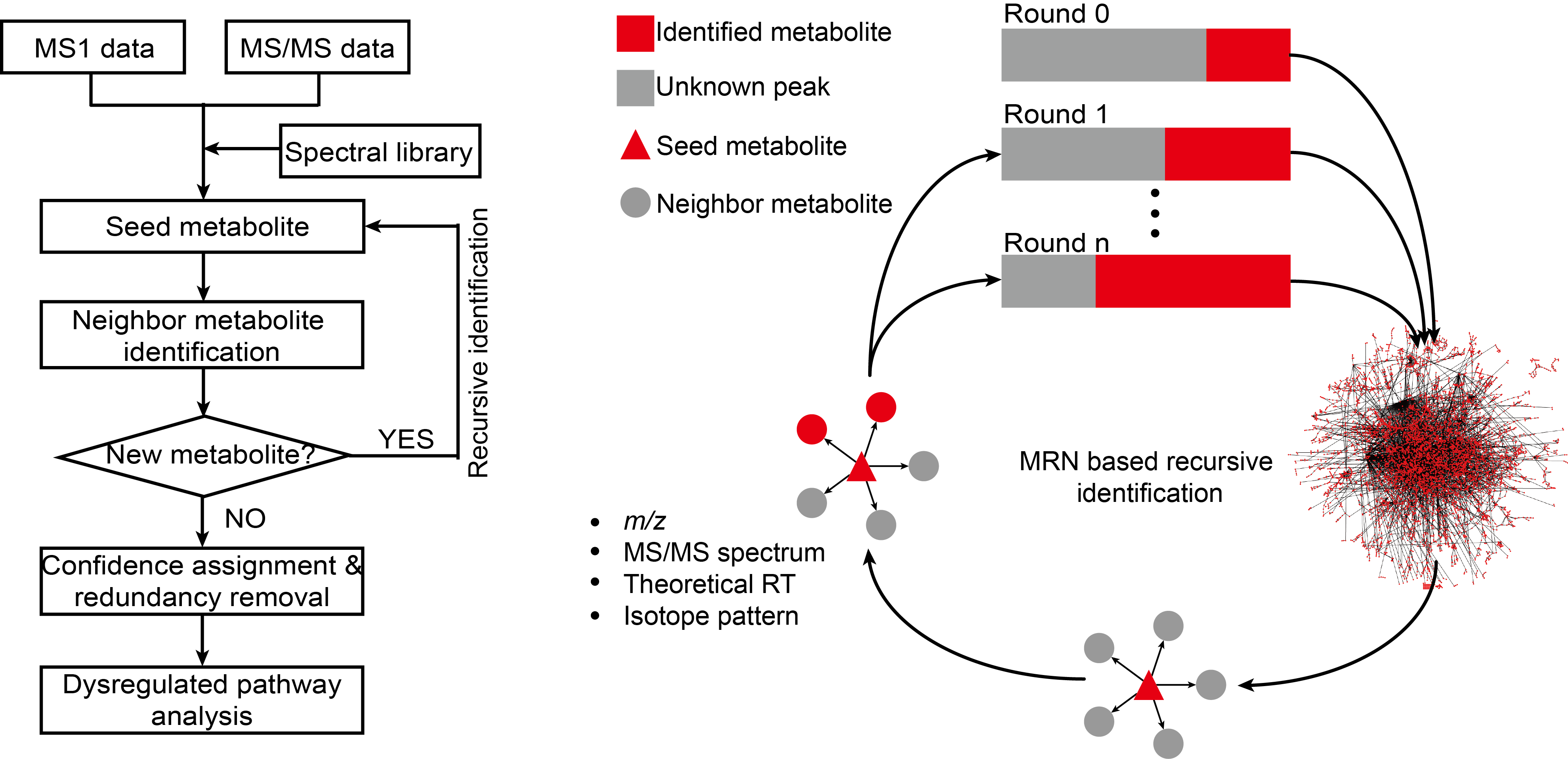
估计阅读时长: 6 分钟访问在线服务: http://metdna.zhulab.cn/ Metabolite identification is the long-standing challenge for liquid chromatography-mass spectrometry (LC-MS)-based untargeted metabolomics. Here, […]
估计阅读时长: 5 分钟目前我们根据质谱数据进行代谢物ROI注释分析,很大一部分的工作是建立在已经可以被纯化的化合物的纯标准品所建立的标准品库数据的比对操作之上的。但是依赖于质谱参考谱图数据库所完成的代谢物注释分析,也仅能够得到很小的一部分结果,因为能够纯化或者合成的化合物在整个自然界中目前只占比较小的一部分。并且购买标准品也会需要耗费大量的实验室资金预算。 Order by Date Name Attachments The-Periodic-Table • 2 MB • 887 click 2022年3月20日Leucine[M+H]+ • 33 […]

估计阅读时长: 10 分钟https://github.com/xieguigang/ms-imaging Order by Date Name Attachments HR2MSI_mouse_urinary_bladder_S096_RGB • 7 MB • 936 click 2021年11月13日peerj-cs-07-585 • 16 […]
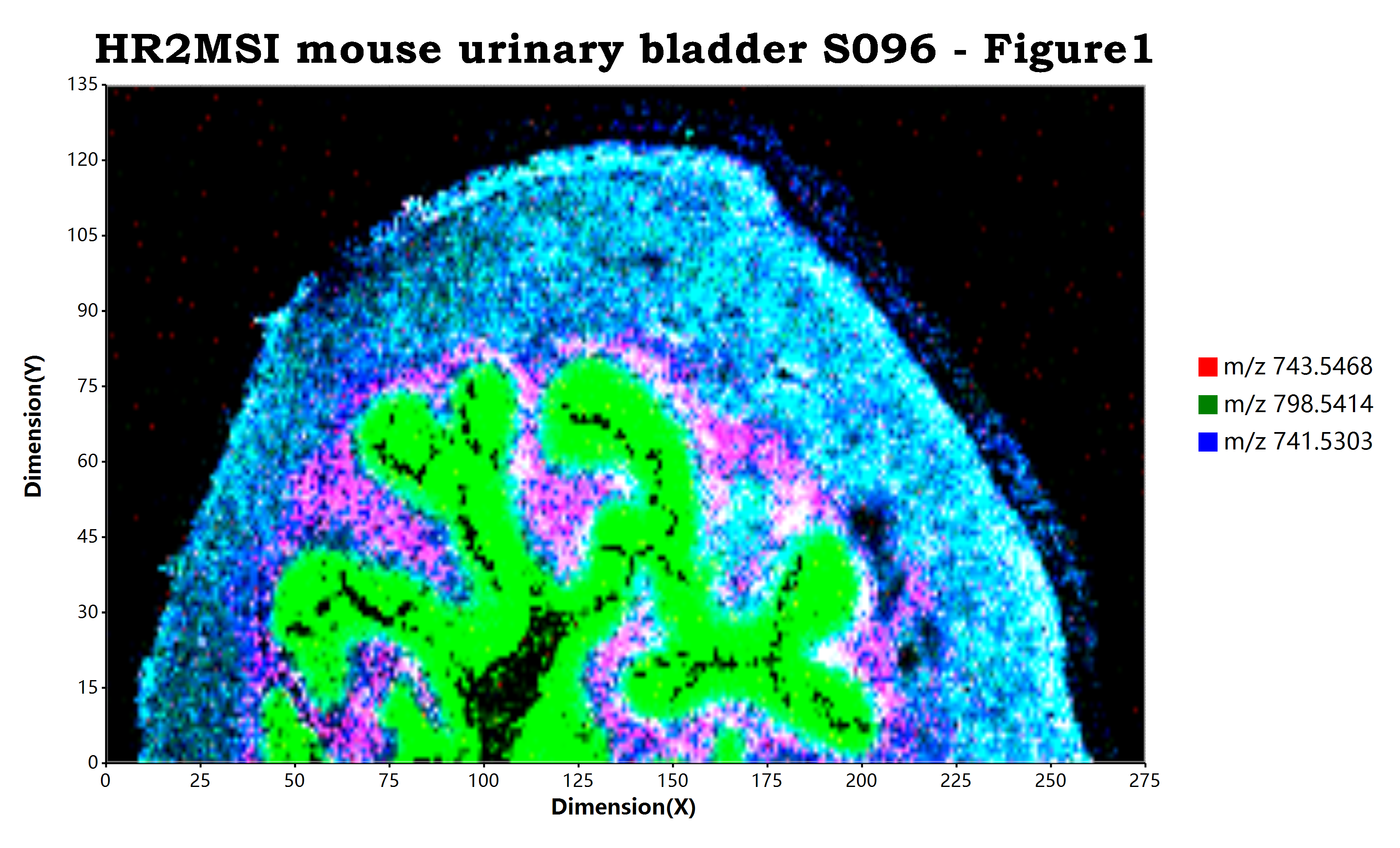
估计阅读时长: 11 分钟https://github.com/xieguigang/ms-imaging Mass spectrometry imaging ( MSI) is a technique used in mass spectrometry to visualize the […]
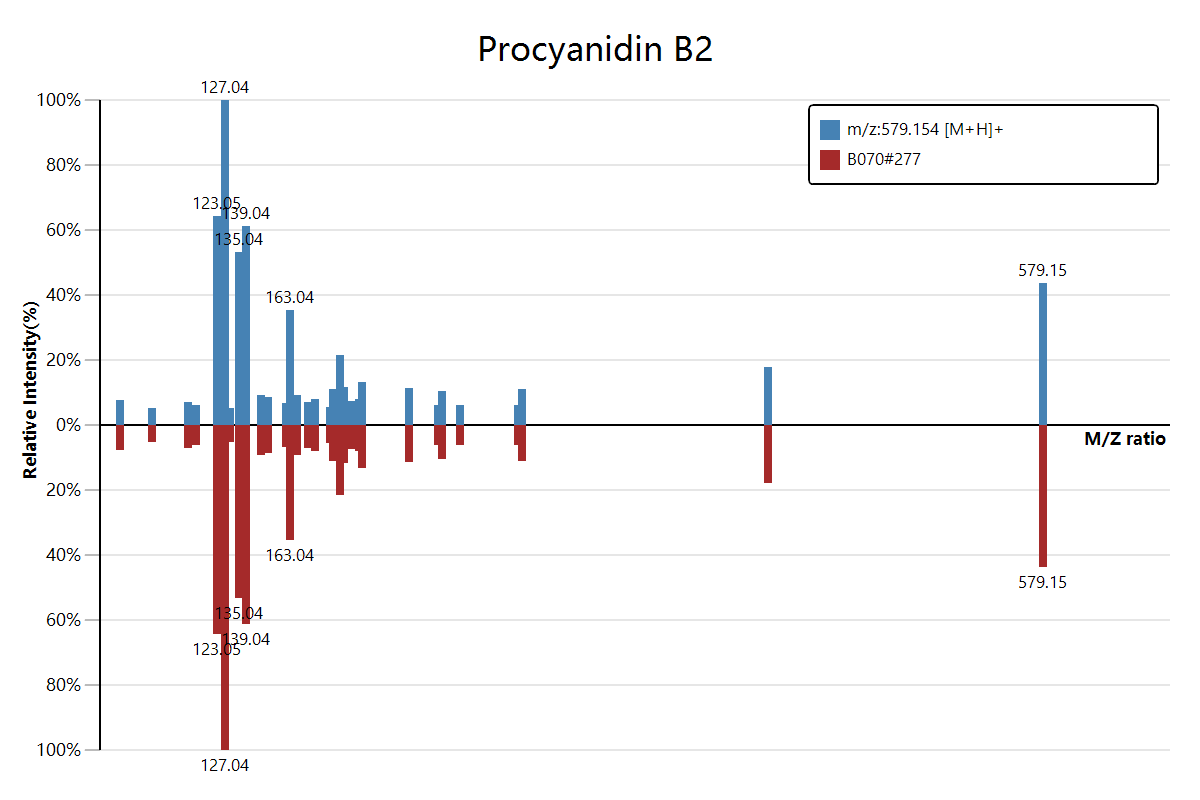
估计阅读时长: 2 分钟https://mzkit.org/ 代谢组文章整个坎坷的发表经历中,大家可能都会遇到的一个老大难问题就是我们有时候会需要从原始数据中得到物质注释结果的二级质谱图数据。对于熟悉xcms程序包的同学,获取二级质谱图可能会比较容易:无非就是加载原始数据,然后按照m/z和rt找到对应的二级scan就好了。但是,这种方法会需要编写脚本来完成。 Order by Date Name Attachments download • 33 kB • 864 click 2021年8月4日[278][MS_MS] FTMS […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?