
文章阅读目录大纲
在BILIBILI上观看视频:《【BioNovoGene Mzkit教程】代谢组学原始数据处理基础》
最近我在B站的视频页面下发现了这样的一条评论,面对质谱数据分析领域内的初学者的求教,其实自己也是非常的诚惶诚恐的。因为在视频中所使用的脚本语言是自己开发的一门新语言,所以可能给一些初学者造成了一部分的困扰哈哈😅😄😅😅。首先先对这个粉丝说一声抱歉哈。

针对上述的提问,我的回答大概是有以下的几点:
- 在B站的教程视频《【BioNovoGene Mzkit教程】代谢组学原始数据处理基础》中,所提到的脚本语言其实并不是真正的R语言,而是基于R语言的向量化编程特性而仿照着开发出来的一门国产编程语言
R# - 所以
netCDF.utils - 因为R#语言的开发结合了多种语言的特性,所以看起来可能会有一些四不像。但是脚本中所展示的
importsusing相关的代码语法其实是来自于C#语言或者VB.NET语言之中的using。
那说完了上面的针对评论回复的几点,下面我们开始进行一些拓展性的知识讲解吧:关于如何使用上R#语言进行质谱数据分析。
首先,先向大伙简单介绍一下MZKit桌面工作站软件的一些背景信息。在视频中所介绍的MZKit软件,其实是帕诺米克从2017年启动的一个质谱原始数据开源项目,相关的项目源代码大家可以从Github上下载得到:https://github.com/xieguigang/mzkit
目前呢,经过多年的发展,MZKit开源项目目前在质谱原始数据处理相关领域内的应用其实已经比较广泛了:就帕诺米克而言,MZKit软件以及R#
既然R#

首先,通过软件左上角的【File】菜单入口打开软件的主菜单;接着可以在主菜单之中看见有【MZKit Data Toolkit】菜单栏;就是在这个菜单栏之中,展开后可以发现有一个命令行图标的东西【Developer CommandLine Tools】;对,就是它了,点击这个东西我们就可以进入到R#

那现在我们来编写一个进行CDF数据可视化的脚本来初步讲解MZKit软件自带的质谱数据处理函数库的使用吧。
像传统的R语言编程一样,一般编程语言环境之中是不包含有针对具体的实际问题解决工具的。所以在进行相关的实际问题处理之前,我们需要导入面向具体实际问题进行数据处理的相关的软件包。在这里我们先进行接下来所需要解决的具体问题的描述:通过读取CDF文件之中的色谱图数据,然后分别通过ggplot软件包或者mzkit软件包绘制出来。
那根据上面的问题描述,实际上我们就可以分解出三个脚本操作相关的模块:CDF文件数据读取模块,ggplot绘图模块或者mzkit质谱数据可视化模块。那么首先,我们向脚本执行环境之中导入CDF文件数据读取模块netCDF.utils
CDF文件数据读取
imports "netCDF.utils" from "base";
具体的来讲,上面的语句其实就是从base.dll
- open.netCDF: 按照一个所设定的文件路径,打开CDF文件流
- var: 从CDF文件流之中获取得到指定名称的变量信息
- getValue: 基于特定的变量信息读取CDF文件之中所存储的向量数据
对于气质色谱数据分析而言,CDF文件是一种比较通用的标准格式,CDF文件里面主要是存储很多个向量数据,而气质色谱的总离子流相关的向量变量信息在CDF文件中分别为:
- scan_acquisition_time 向量即为总离子流图的保留时间向量信息
- total_intensity 顾名思义就是总离子流图的每一个保留时间点上所对应的总离子信号响应强度信息
那,现在了解到了上面的主要相关信息之后,我们就可以写出如下脚本进行数据的读取了:
using cdf as open.netCDF(file = "C:\\Program Files\\BioNovoGene\\mzkit_win32\\demo\\200ppm-scan.CDF") {
const readVector = function(name) {
const varinfo = var(cdf, name = name);
const vector = getValue(varinfo);
vector;
}
const scan_time = readVector("scan_acquisition_time");
const totalIons = readVector("total_intensity");
const TIC = data.frame(scan_time, totalIons);
print(TIC, max.print = 13);
}现在保存一下我们的脚本文件,在命令行窗口中输入R#;接着呢,复制文件路径,然后粘贴我们的文件路径在命令行输出窗口中,例如像下面这样子:

R# C:\Users\Administrator\Desktop\plot_cdf_tic.R最起始的R#

ggplot绘制总离子流色谱图
现在,我们已经拥有了绘图相关的数据,那现在我们可以做绘图操作了。首先我们祭出R语言的绘图利器,首先展示一下在R#编程语言环境之中通过ggplot进行总离子流色谱图的绘制:
require(ggplot);
bitmap(file = `${@dir}/tic_ggplot.png`, width = 2400, height = 1440) {
ggplot(TIC, aes(x = "scan_time", y = "totalIons"), padding = "padding: 200px 150px 200px 300px;")
+ geom_line(width = 7)
+ ggtitle("Total Ions Chromatographic(TIC)")
+ scale_x_continuous(labels = "F2")
+ scale_y_continuous(labels = "G2")
;
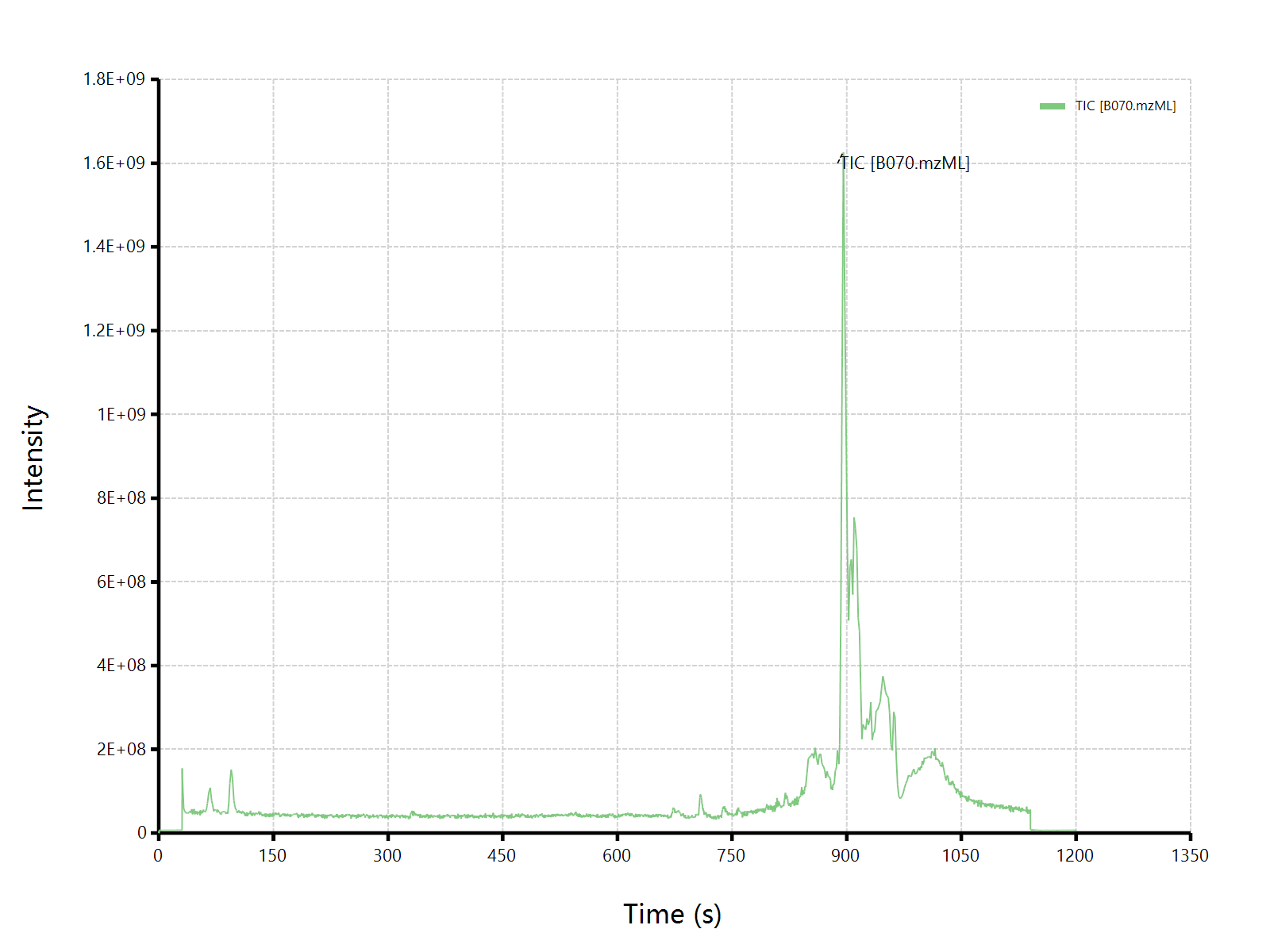
}通过执行上面的一段ggplot的绘图脚本代码,我们得到了如下所示的TIC图数据可视化输出结果:

MZKit程序包绘制总离子流色谱图
上面我们展示的是基于ggplot程序包按照一般的数据绘制流程做的一个数据可视化,那现在假若我们想要基于MZKit程序包进行上面所展示的类似的TIC色谱图的绘制操作呢?当然也是没问题的了,因为MZKit软件包就是专门进行质谱数据处理操作的工作,绘制这种基础原始数据图当然不在话下了,首先来看绘图代码:
require(mzkit);
#' A chromatogram is the trace generated by the detector
imports "chromatogram" from "mzkit";
imports "visual" from "mzplot";
tic = toChromatogram(as.chromatogram(TIC$scan_time, TIC$totalIons));
bitmap(file = `${@dir}/tic_mzkit.png`, width = 2400, height = 1440) {
plot(chromatogram::overlaps(list(demo_data = tic)), show.labels = FALSE);
}从上面的绘图代码可以看见,相关的绘图操作和ggplot相比较,自定义方面的内容(例如坐标轴,标题之类的信息)会较少一些,但是相关的绘图操作却简化了一些:我们只需要通过chromatogram对象相关的操作函数,将读取出来的原始数据转换为chromatogram对象,接下来就可以直接通过plot函数绘制出来了!除了操作简单这个特点,默认输出的图形样式也要漂亮了许多。

下面截图的是在这篇文章中所展示的基础数据作图功能的全部代码了:

上面的所有代码都是基于MZKit桌面工作站软件最新发布的版本所完成的,如果各位读者需要更新或者安装最新版本的MZKit桌面工作站软件,可以访问BioDeep网站进行下载:http://www.biodeep.cn/downloads?lang=zh-CN
希望这篇评论回复文章可以解开上面的粉丝的一部分疑惑
- 单细胞视角下的微生物基因组代谢酶嵌入分析 - 2026年2月25日
- 基因组代谢酶层级嵌入 - 2026年2月23日
- 基因组功能吉布斯LDA主题建模 - 2026年2月23日

No responses yet