
文章阅读目录大纲

基于UMAP工具进行简单的自动化组织分区操作
在这里我们假设已经可以正常的将空间代谢数据导入至MZKit工作站软件之中。假若需要借助于MZKit工作站软件进行切片组织样本的自动化分区操作,相关的功能可以在【MSI Analysis】菜单栏中寻找到。在这里我们打开【Show Map Layer】按钮,选择【UMAP and clustering】功能。

基于降维的组织自动化分区原理
因为降维操作一般是一种特征提取操作,所以经过降维之后,在高维度空间上无法显现的特征,在低维度会呈现出来。在高维度空间散落的相近的数据点,在经过特征提取之后,低维度上会产生相似的特征信息,相互聚集在一簇。这样子我们就可以在低维度空间上通过一些聚类算法讲这些特征进行聚类,最后将聚类特征结果标记到各个散点上的对应的原始成像空间上,我们就可以看见组织分区的结果了。

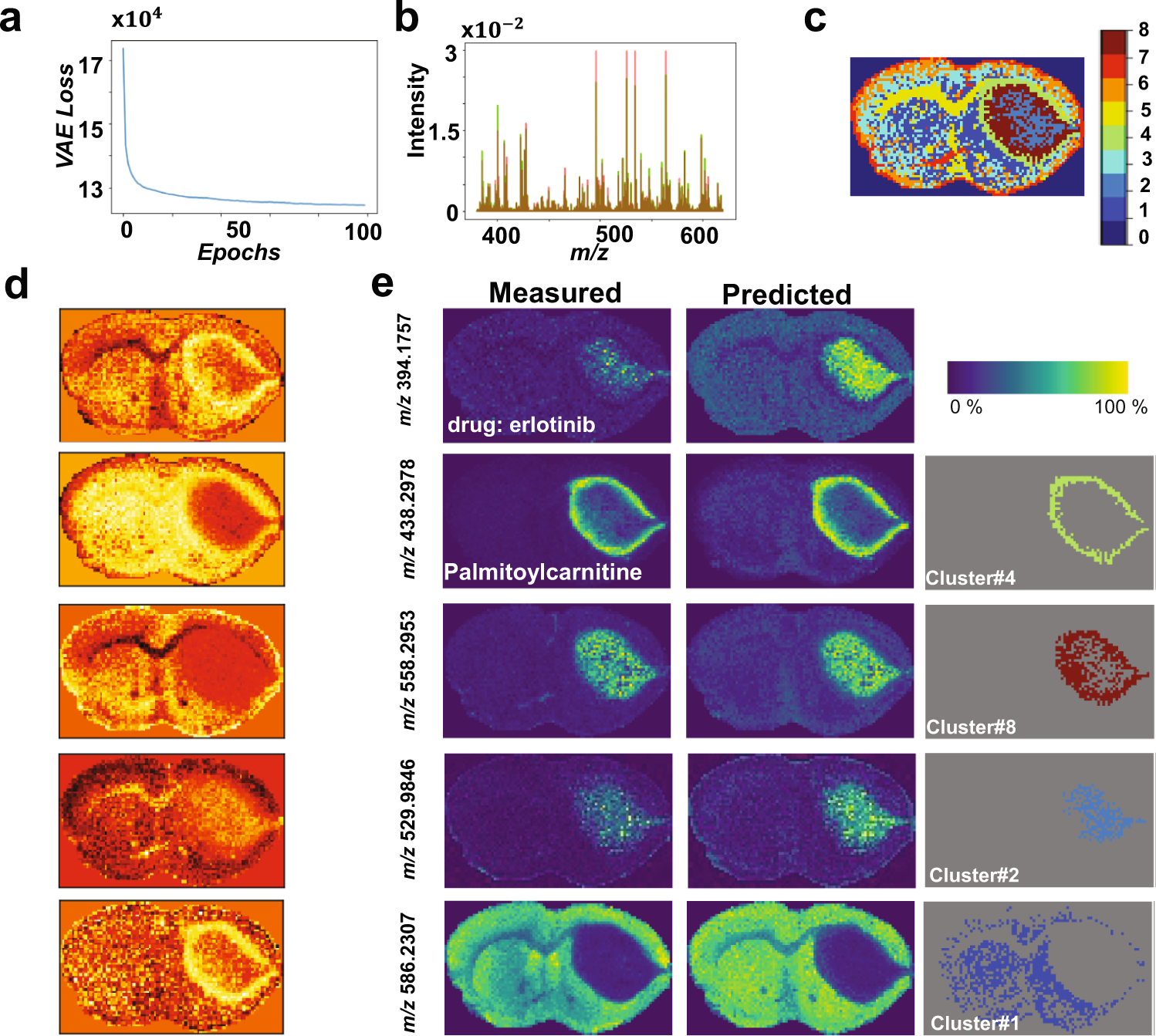
Fig. 3: Analysis of 2D MALDI FT-ICR MSI dataset of PDX mouse brain model of glioblastoma.
Abdelmoula, W.M., Lopez, B.GC., Randall, E.C. et al. Peak learning of mass spectrometry imaging data using artificial neural networks. Nat Commun 12, 5544 (2021). https://doi.org/10.1038/s41467-021-25744-8
在这里我们使用Nature上的一篇方法学文章里面的结果数据进行复现。
操作步骤1:提取空间表达矩阵
在进行组织的自动化分区操作之前,MZKit工作站软件首先会需要获取得到一个空间矩阵数据,然后基于我们所创建的高维度的空间矩阵数据进行降维处理。在MZkit软件之中会存在有数据缓存工作机制。假设是第一次进行当前样本文件的分析处理的话,一般会跳出下面所示的对话框:

在这个对话框中,我们需要设定质谱数据处理相关的参数信息,进行空间表达矩阵的导出:
- mzdiff,即分子质量误差,与转录组相比较,基因一般可以通过一个id编号进行一一对应的表示,但是在质谱数据分析中,一个分子对象的描述,一般是以一个在一定误差范围内的m/z数字来代表的。在这里设定的即为描述这个mz误差范围的分子质量误差。在这里推荐使用软件的默认值0.5道尔顿
- intocutoff,代谢物离子响应度过滤阈值。这个参数是一个百分比参数,值位于0到1之间,表示0%到100%,按照文献推荐,在这里软件默认将5%以下的数据看作为噪声数据进行过滤操作
- TrIQ,这个参数用于削平离子信号响应的最高值,减少极值差异,参数值位于0到1之间,1表示不进行最大值削平操作,一般不建议将这个参数值设置低于0.8,在这里推荐使用软件的默认参数1.
在等待软件完成矩阵数据的导出之后,软件会自动打开【UMAP Tool】分析工具。

在【UMAP Tool】分析工具中,大致分为两大部分:
- Manifold 模块用于配置参数进行原始的高维度空间表达矩阵进行降维处理
- Clustering 模块用于配置参数,基于降维结果进行聚类分析
在本分析工具之中,需要经过的操作步骤为:【Manifold】->【Clustering】->【Save】将聚类结果应用到空间样本上。
操作步骤2:数据降维

在这里主要是基于UMAP方法进行数据降维操作,在这里推荐直接使用软件的默认参数进行分析。相关的方法参数的含义,可以阅读这篇帮助文档:https://umap-learn.readthedocs.io/en/latest/api.html。在这里我们只需要设定好UMAP的参数之后吗,点击【Run】按钮执行分析命令即可。
操作步骤3:聚类分析
在MZkit工作站软件之中,目前提供了3种聚类方法用于自动化组织分区操作。我们均可以通过调整这三种方法的相关参数来进行文献中的分区结果的复现。
a. K-Means方法
K-Means方法是我们最容易理解的一种聚类方法,其依照欧几里得距离将数据点按照我们期望的聚类数量将我们的数据划分为不同的类别。在软件中,我们只需要拖动滑动条,设置期望的聚类结果数量,然后执行【Set K-Means】命令即可

b. DBSCAN方法
因为K-Means是一种基于数据点与中心点的欧几里得距离进行聚类的操作,所以K-Means一般是会得到一种球形的区域为聚类结果。假设我们观察UMAP散点图的聚类染色结果情况,一般很有可能会发现因为球形区域的计算,不同散点簇上的数据点可能会被K-Means分配为相同的颜色。DBSCAN方法是一种可以解决这种球形聚类的缺点的聚类方法。DBSCAN方法采用类似于KNN的原理,基于密度阈值进行任意空间形状的聚类。

在这里我们需要调整两个参数值:
- Min Points参数,类似于KNN的K值
- Eps参数,是用于切断两个聚类簇的密度阈值,具体的密度阈值与具体的数据分布有关
c. Graph图聚类方法
图聚类方法是一种基于树图构建的聚类方法。在进行树图构建的过程中,假设某一个数据点其和当前的树节点的相似度超过一个阈值,则会将目标数据点划分到当前的树节点中,从而实现将数据进行聚类的操作。

在这里,我们只需要在软件界面上设定好这个相似度阈值即可
操作步骤4:结果数据导出
假设需要将降维和聚类的结果进行导出,自行绘制相关的散点图可视化的话,可以直接点击【Download Result】链接,即可从软件上下载我们的结果数据表格,将这个数据表格中的内容导入到R或者Python环境中,进行后续的下游绘图可视化。

在我们所导出的表格中,其格式为:
a. 第一列数据:为空间代谢数据的斑点(x,y)空间坐标信息
b. 第二列class列为数据点的聚类结果
c. 第3,4,5列则是UMAP降维的前三个维度信息,可以基于这三个维度信息或者前两个维度信息进行相关的散点图绘制

操作步骤5:保存自动化组织分区结果
最后,完成了上面的操作后,我们只需要要点击【Save】按钮,将我们的聚类结果数据保存到空间代谢数据分析工作区中,即可查看到分区的可视化结果。如果需要将分区结果继续保存为文件,可以通过MZKit工作站的【Export Region】菜单进行保存。

- 单细胞视角下的微生物基因组代谢酶嵌入分析 - 2026年2月25日
- 基因组代谢酶层级嵌入 - 2026年2月23日
- 基因组功能吉布斯LDA主题建模 - 2026年2月23日

One response
This clarifies everything perfectly.