
文章阅读目录大纲
给定一组n个字符串数组,找到包含给定集合中每个字符串的最小字符串作为子字符串。我们可以假设这个字符串数组中没有字符串是另一个字符串的子字符串。那么基于上面的描述,我们就可以得到下面所示的问题求解目标:
let arr[] = ["catg", "ctaagt", "gcta", "ttca", "atgcatc"]
// output: gctaagttcatgcatc上面的问题描述实际上是一个最短超字符串问题(shortest common superstring)
SCS算法实现
为了求解SCS问题,最简单的算法是贪心算法,既简单的通过字符串之间的重叠位置进行公共字符串的拼接,既可以得到最短的公共超字符串计算结果:
Let arr[] be given set of strings.
1) Create an auxiliary array of strings, temp[]. Copy contents
of arr[] to temp[]
2) While temp[] contains more than one strings
a) Find the most overlapping string pair in temp[]. Let this
pair be 'a' and 'b'.
b) Replace 'a' and 'b' with the string obtained after combining
them.
3) The only string left in temp[] is the result, return it.基于上面的算法描述,我们可以编写出下面的贪心算法实现代码:
<Extension>
Public Function ShortestCommonSuperString(strs As IEnumerable(Of String)) As String()
Dim seqs As String() = strs.ToArray
Dim l As Integer = seqs.Length
Dim p As Integer
Dim q As Integer
Dim finalStr As String
Do While l > 1
p = -1
q = -1
finalStr = runIteration(l, seqs, p, q)
l -= 1
seqs(p) = finalStr
seqs(q) = seqs(l)
Loop
Return seqs
End Function
Private Function runIteration(l As Integer, seqs As String(), ByRef p%, ByRef q%) As String
Dim currMax As Integer = Integer.MinValue
Dim finalStr As String = Nothing
For j As Integer = 0 To l - 1
For k As Integer = j + 1 To l - 1
Dim str As String = seqs(j)
Dim b As String = seqs(k)
If str.Contains(b) Then
If b.Length > currMax Then
finalStr = str
currMax = b.Length
p = j
q = k
End If
ElseIf b.Contains(str) Then
If str.Length > currMax Then
finalStr = b
currMax = str.Length
p = j
q = k
End If
Else
' find max common prefix and suffix
Dim maxPrefixMatch = MaxPrefixLength(str, b)
If maxPrefixMatch > currMax Then
finalStr = str & b.Substring(maxPrefixMatch)
currMax = maxPrefixMatch
p = j
q = k
End If
Dim maxSuffixMatch = MaxPrefixLength(b, str)
If maxSuffixMatch > currMax Then
finalStr = b & str.Substring(maxSuffixMatch)
currMax = maxSuffixMatch
p = j
q = k
End If
End If
Next
Next
Return finalStr
End Function
Private Function MaxPrefixLength(a As String, b As String) As Integer
Dim max As Integer = 0
Dim prefix$
For i As Integer = 0 To b.Length - 1
prefix = b.Substring(0, i + 1)
If a.EndsWith(prefix) Then
max = i + 1
End If
Next
Return max
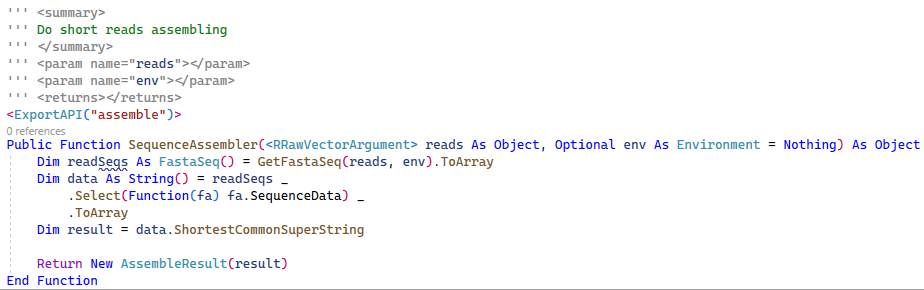
End Function在目前测序数据分析流程之中,因为技术的限制,往往只能够得到一些目标序列片段。所以我们首先会需要将比较短的测序reads结果按照一定的方式组装成一条比较长的序列结果,在这个过程之中,实际上就与SCS问题非常的相似了。我们可以基于SCS算法实现出一个最简单的测序reads组装算法。

FastQ文件
说到了RNA-seq测序数据分析,在这里再顺便介绍一下FastQ文件相关的信息。
FastQ文件的格式比FastA文件格式要复杂一些:除了对应的序列标题头和序列数据本体外,在FastQ文件之中还额外的增加了测序的质量数据。下面展示了简单的FastA与FastQ的格式对比:
-----FastA-----
> SRR031716.1 HWI-EAS299_4_30M2BAAXX:3:1:944:1798 length=37
GTGGATATGGATATCCAAATTATATTTGCATAATTTG
-----FastQ-----
@SRR031716.1 HWI-EAS299_4_30M2BAAXX:3:1:944:1798 length=37
GTGGATATGGATATCCAAATTATATTTGCATAATTTG
+SRR031716.1 HWI-EAS299_4_30M2BAAXX:3:1:944:1798 length=37
IIIIIIIIIIIIIIIIIIIIIIIIIIIII8IIIIIII可以看得见在FASTQ之中包含了每一个读长最原始的信息,通常每4行来描述一个读长。
- 第一行:以@开头,然后是一串和测序过程相关的信息
- 第二行:具体的ACGT核酸序列
- 第三行:以+号开头,和第一行相似
- 第四行:一串字符组合,每一个字符代表一个碱基的质量评分,所以该行的长度应该和第二行碱基的长度是一致的。
继续阅读:二代测序质量控制(FastQC)
测序质量得分计算
| Phred Quality Score | Probability of incorrect base call | Base call accuracy |
|---|---|---|
| 10 | 1 in 10 | 90% |
| 20 | 1 in 100 | 99% |
| 30 | 1 in 1000 | 99.9% |
| 40 | 1 in 10,000 | 99.99% |
| 50 | 1 in 100,000 | 99.999% |
| 60 | 1 in 1,000,000 | 99.9999% |
对于在FastQ文件之中的第四行测序质量信息,其长度与序列的长度一致,每一个得分字符与短reads序列之中的字符也一一对应。FastQC得分(Phred quality score)分值越高,表示对应的碱基的准确性越好。关于质量得分的计算,可以通过取出对应的字符的ASCII码值减掉33即可:
''' <summary>
''' The character '!' represents the lowest quality while '~' is the highest.
''' Here are the quality value characters in left-to-right increasing order
''' of quality (ASCII):
''' </summary>
''' <remarks></remarks>
Public Const QualityOrders$ = "!""#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~"
''' <summary>
''' 测序质量,每个字符对应第2行每个碱基,第四行每个字符对应的ASCII值减去33,
''' 即为该碱基的测序质量值,比如@对应的ASCII值为64,那么其对应的碱基质量值是31。
''' 从Illumina GA Pipeline v1.8开始(目前为v1.9),碱基质量值范围为0到41。
''' </summary>
''' <param name="q"></param>
''' <returns></returns>
Public Shared Iterator Function GetQualityOrder(q As String) As IEnumerable(Of Integer)
For Each c As Char In q
Yield Asc(q) - 33
Next
End Function如果想要调用上面的代码进行fastQ文件之中的序列质量计算,可以通过下面的代码进行计算函数的调用:
FQ.FastQ _
.GetQualityOrder(DirectCast(q, FQ.FastQ).Quality) _
.Select(Function(d) CDbl(d)) _
.ToArrayJavascript调用实例
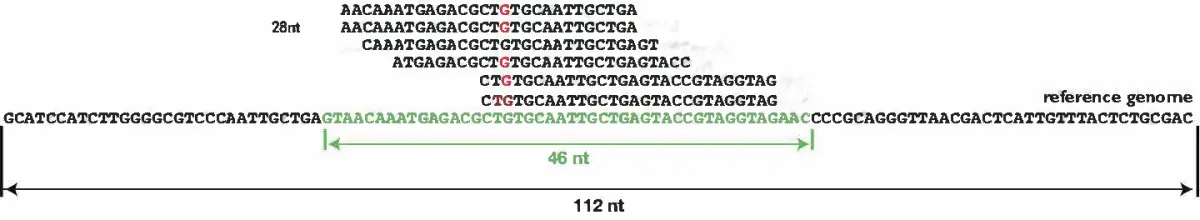
使用VB.NET将SCS算法封装成了一个函数用于javascript外部调用,下面的一段javascript代码展示了基于SCS
require("GCModeller");
// load the fastq module from rna-seq package
// inside the GCModeller
import {FastQ} from "rnaseq";
// do short reads assembling via SCS algorithm
var assem = FastQ.assemble([
"AACAAATGAGACGCTGTGCAATTGCTGA",
"AACAAATGAGACGCTGTGCAATTGCTGA",
"CAAATGAGACGCTGTGCAATTGCTGAGT",
"GAAATGATACGCTGTGCAATTGCTGAGA",
"ATGAGACGCTGTGCAATTGCTGAGTACC",
"CTGTGCAATTGCTGAGTACCGTAGGTAG",
"CTGTGCAATTGCTGAGTACCGTAGGTAG"
])
// view the short reads assemble result
console.table(assem)
- 单细胞视角下的微生物基因组代谢酶嵌入分析 - 2026年2月25日
- 基因组代谢酶层级嵌入 - 2026年2月23日
- 基因组功能吉布斯LDA主题建模 - 2026年2月23日

No responses yet