
文章阅读目录大纲
 http://mzkit.org/
http://mzkit.org/
质谱成像是以质谱技术为基础的成像方法,该方法通过质谱直接扫描生物样品成像,可以在同一张组织切片或组织芯片上同时分析数百种分子的空间分布特征。
今天在这篇文章中,我主要是为大家介绍一下我们是可以怎样使用开源的mzkit软件进行质谱成像可视化操作的。在学习怎样使用mzkit软件进行质谱成像原始数据文件查看之前,我们有必要先来了解一些于质谱成像相关的原始数据文件。
质谱成像原始数据文件有哪些?
在质谱成像领域内,主要包含有下面的三种原始数据格式文件:
- 赛默飞公司的Raw文件格式
- imzML原始数据文件格式
- 帕诺米克公司的mzPack原始数据文件格式

赛默飞Raw文件
对于赛默飞的Raw文件格式我们在这里就不做过多介绍了。在代谢组学领域内基本上会绕不开赛默飞的仪器,我们做非靶向代谢组几乎百分之百都会接触到Raw文件。对于空间代谢组学而言,由于质谱成像的imzML文件文件就是转换格式于最原始的Raw下机原始数据文件;所以理论上我们只需要知道扫描斑点数信息,就可以直接从Raw文件中计算出质谱成像所需要的空间坐标信息。
对于从Raw文件之中计算出空间坐标信息,我们可以使用下面一段公式代码来完成:
Private Function PixelScanId(pixels As Size) As Func(Of SingleScanInfo, Integer, String)
Return Function(scan, n)
If scan.MSLevel = 1 Then
Dim y As Integer = stdNum.Floor(n / pixels.Width) + 1
Dim x As Integer = n - (y - 1) * pixels.Width + 1
Return $"[MS1][Scan_{scan.ScanNumber}][{x},{y}] {scan.FilterText}"
Else
Return $"[MSn][Scan_{scan.ScanNumber}] {scan.FilterText}"
End If
End Function
End FunctionimzML原始数据文件

imzML主要是由两个文件构成:一个数据文件本体,也就是imzML文件了;另一部分就是一个ibd二进制文件,里面专门用于存储质谱矩阵信息。我们看imzML的文件拓展名就可以知道,imzML这个文件格式是专门应用于质谱成像领域的文件格式,因为其文件拓展名为image mzML。对于其数据文件本体而言,其就是一个没有质谱数据的mzML文件;因为其将质谱数据存放于外部的ibd二进制文件,所以读写效率要比文本文件格式的mzML文件要高很多。
帕诺米克公司的mzPack开源原始数据文件格式
mzPack文件有点类似于二进制的mzML文件,其文件内部主要包含了4大区域:质谱扫描数据区域,色谱数据区域,索引区域以及一个Meta元数据信息区域。mzPack文件相比较于赛默飞的Raw文件和XML文本格式的mzXML/mzML/imzML文件都要小非常多,读写效率非常高,因此mzPack目前在帕诺米克公司内部被广泛的应用于非靶向原始数据自动化处理流程之中。
在mzPack文件之中,存在着于imzML类似的空间坐标信息,存储于Meta元素据区域,通过索引号于质谱扫描一一对应。对于mzPack文件格式而言,其也是一种非常优秀的质谱成像原始数据文件格式。
mzkit查看质谱成像原始数据文件
在Mzkit软件之中查看质谱成像的步骤非常的简单。我们一般是加载完数据之后,设定一下目标离子就可以进行成像了。下面我就来介绍一下mzkit中质谱成像的几个操作步骤。
mzkit质谱成像操作步骤
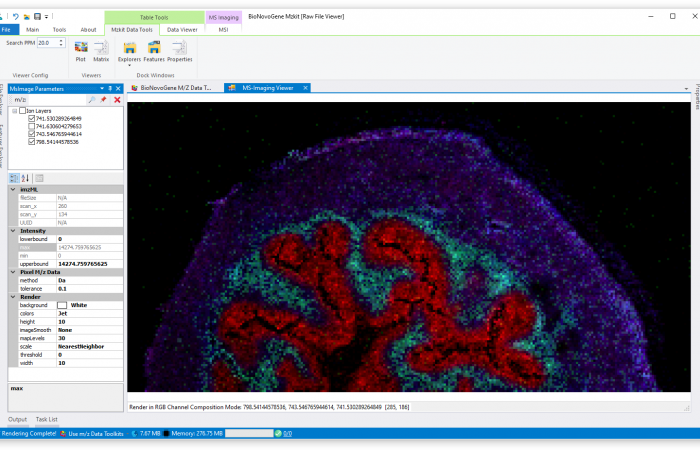
- 在我们打开了mzkit软件之后,我们一般首先可以从【Mzkit Data Tools】菜单栏之中找到质谱成像浏览器,例如:

- 之后,点击它,就可以打开Mzkit的质谱成像浏览器界面了。在最开始的时候,这个浏览器界面是空白的,如下所示:
![]()
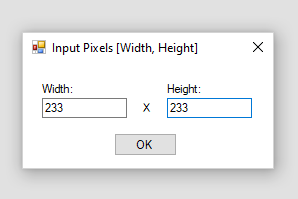
- 接着呢,我们就可以通过点击左上方的文件夹打开一个imzML文件或者赛默飞的Raw文件了。对于imzML文件,因为文件里面已经存在有坐标信息了,所以我们可以直接打开加载目标文件;但是对于赛默飞的Raw文件,我们在打开文件后,加载数据之前还会弹出一个对话框让我们输入扫描点数量信息,我们填完之后就可以正常加载数据了。
国际上通用的MSI的原始数据文件名的编码标准:
因为对于Raw文件而言,我们是需要换算才可以知道空间坐标信息的。而这个换算过程是必须要知道扫描斑点数的信息。所以假若我们通过Raw文件发表我们的结果原始数据,我们必需要将扫描信息编码进入文件名中,其他的同行才可以根据文件名中所提供的信息正确的查看我们的原始数据文件。一般而言,一个MSI原始数据文件的文件名一般为:<物种名/样本来源>_设备信息_像素宽度x高度_空间分辨率_mz范围
例如: MPIBremen_Bputeoserpentis_MALDI-FISH_DHB_233x233pixel_3um_mz400-1200_240k@200
在加载原始文件之后,我们就可以选择离子进行质谱成像渲染了。
- 我们会需要通过左上角的输入框输入我们的目标离子。例如,在这里我们输入目标离子741.5307。之后点击旁边的放大镜的图标,程序就会开始依照我们所设定的目标mz进行原始数据读取操作了。
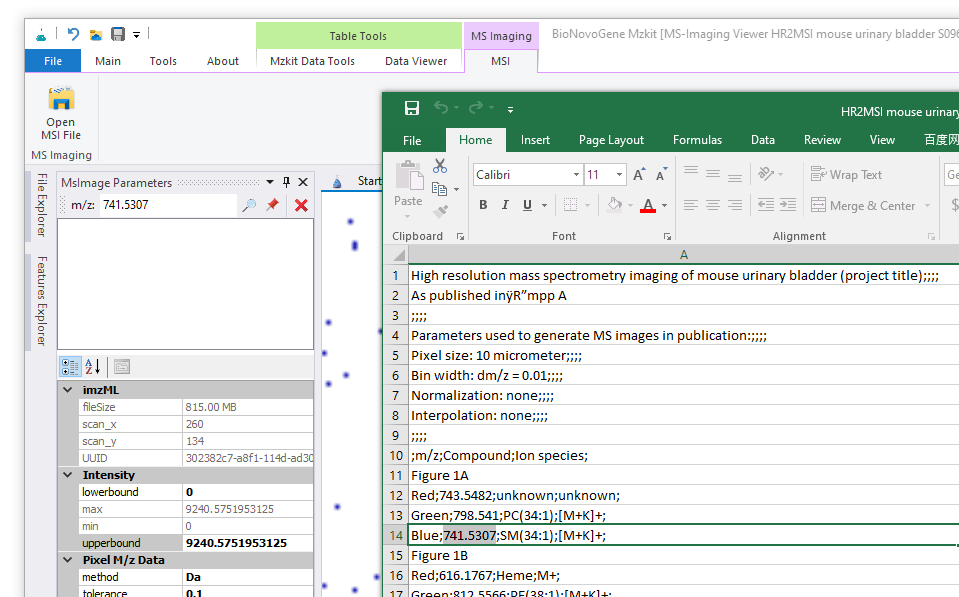
对于通过离子的mz进行原始数据读取,我们还需要设置一个误差窗口。Mzkit软件默认的误差窗口为0.1Da;大家也可以修改这个误差窗口为ppm误差。误差窗口的值的设定,大家可以通过左下角Pixel M/z Data属性输入框进行设置。
- 细心的大家,可能会发现在放大镜旁边还有一个图钉图标。这个图标的作用就是可以让程序暂时记住我们输入的离子。我们一般可以通过这个功能,为mzkit软件提供多个m/z值,从而进行多个离子的质谱成像的渲染工作。如果需要进行多个离子的质谱成像渲染,我们只需要添加好离子列表之后,点击鼠标右键,选择对应的渲染操作菜单就好了。
另外,如果大家在离子图层选择框中单击鼠标右键的话,会发现右键菜单中存在有两种渲染选项。这两种渲染选项的工作模式有一些差异,下面我来为大家稍微讲解一些:
RGB通道叠加渲染模式
RGB通道叠加渲染模式是一般进行文章发表最常用的质谱成像渲染模式,其渲染的原理就是通过所选定的离子的信号强度分别赋值给RGB这三个通道来产生颜色映射。由于我们的颜色是由RGB这三个颜色通道组成的,所以这种模式最多只能够叠加三种离子。这种渲染模式最大的一个特点就是,对于低信号响应的位置都是黑色,因为RGB三个通道的映射值对于低信号响应的时候都是接近于零的(黑色的RGB值为RGB(0,0,0))。
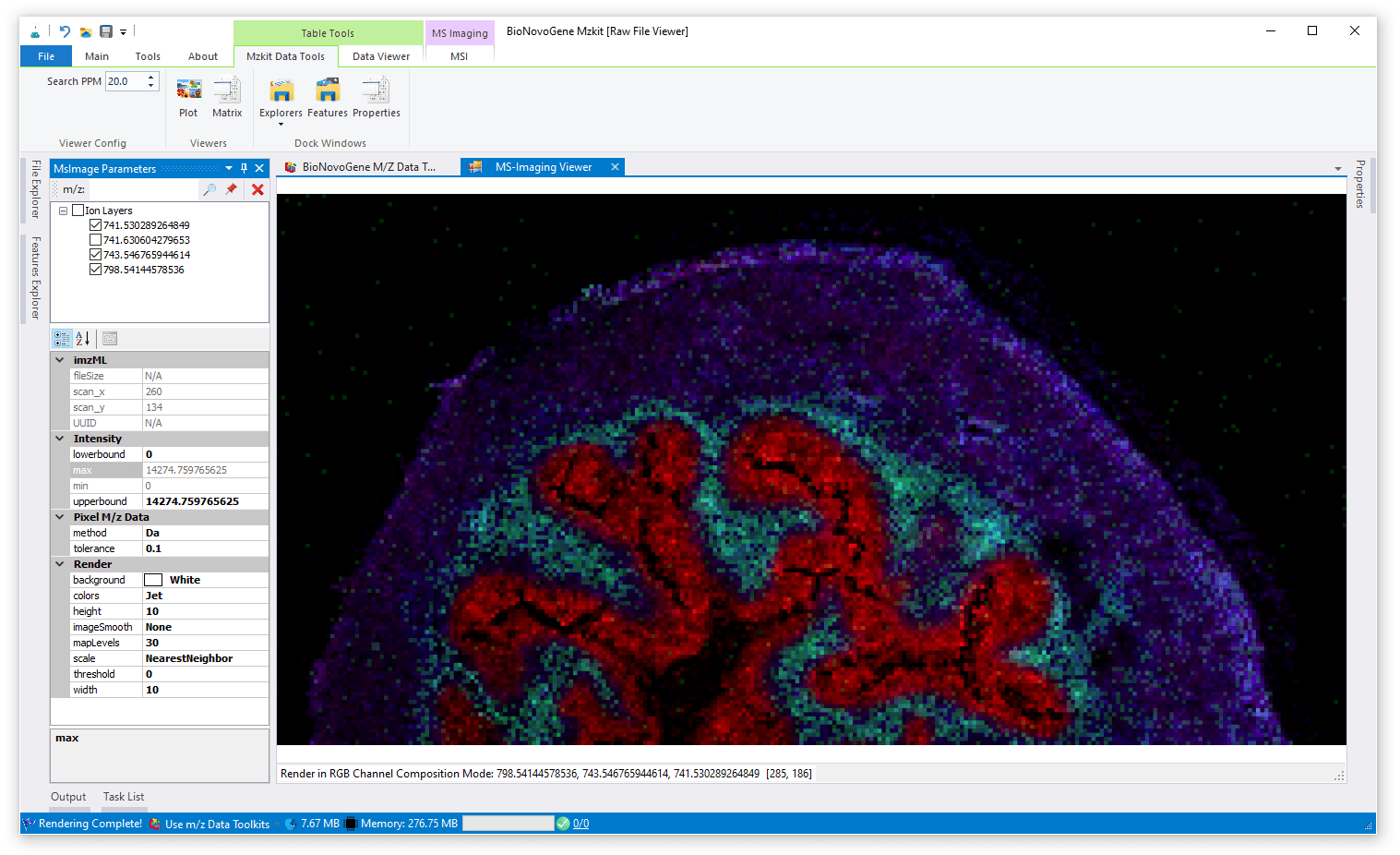
因为低信号响应区域一般而言是要大于高信号响应区域的,所以RGB通道叠加渲染模式的成像结果比较黑
信号混合渲染模式
信号混合渲染模式就是将一个离子或者多个离子的信号分别归一化之后,进行统一缩放,最后映射到一个颜色谱之中产生颜色渲染。其相较于RGB通道叠加渲染,而言,成像结果更加明亮,一般会认为这种渲染模式下的成像图片更加好看。但是一般会因为颜色过多,在多种离子混合渲染的模式下,而无法正常的解释代谢物的表达上的空间分布。信号混合渲染模式对于单个离子的渲染是一种最优的选择。
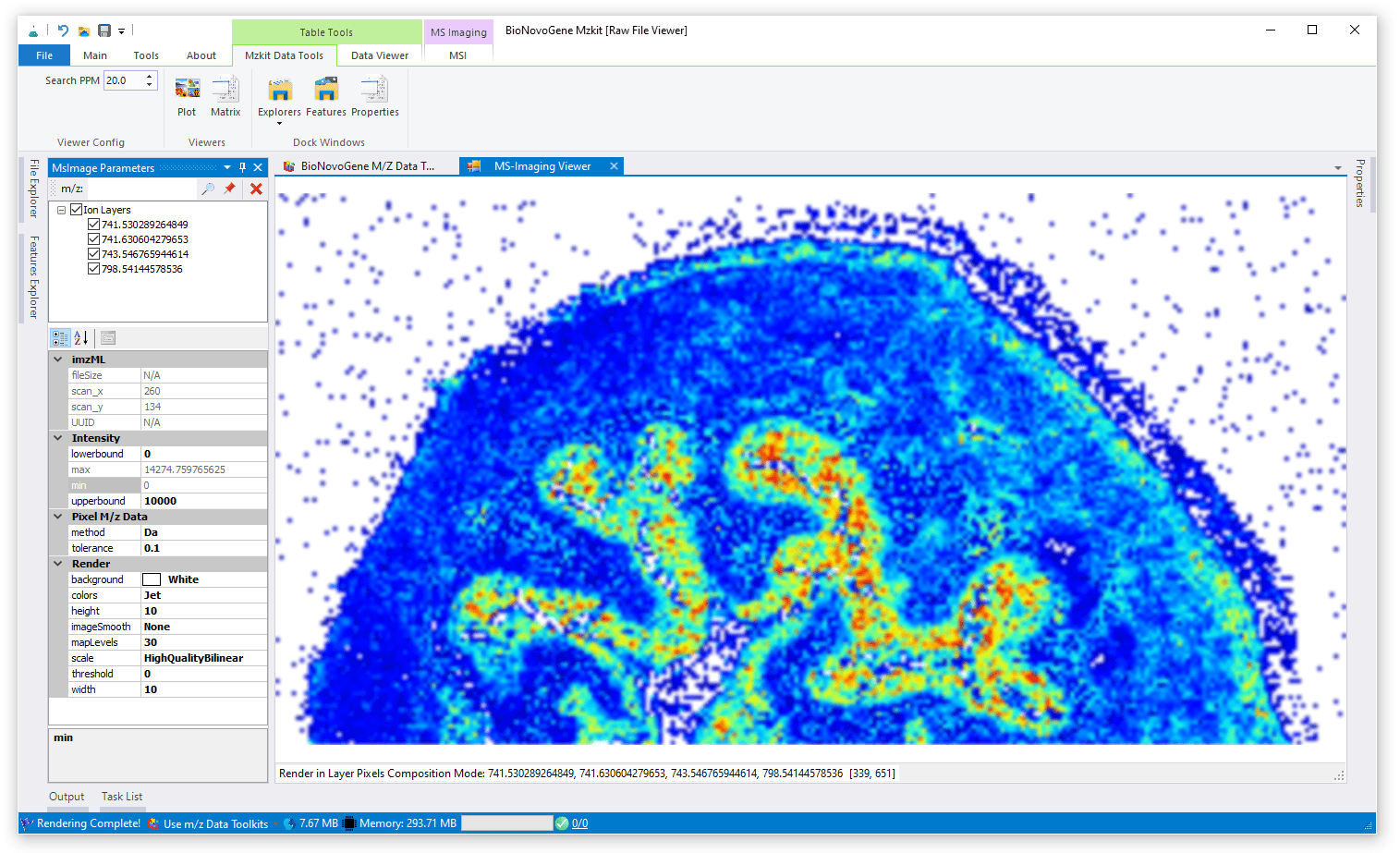
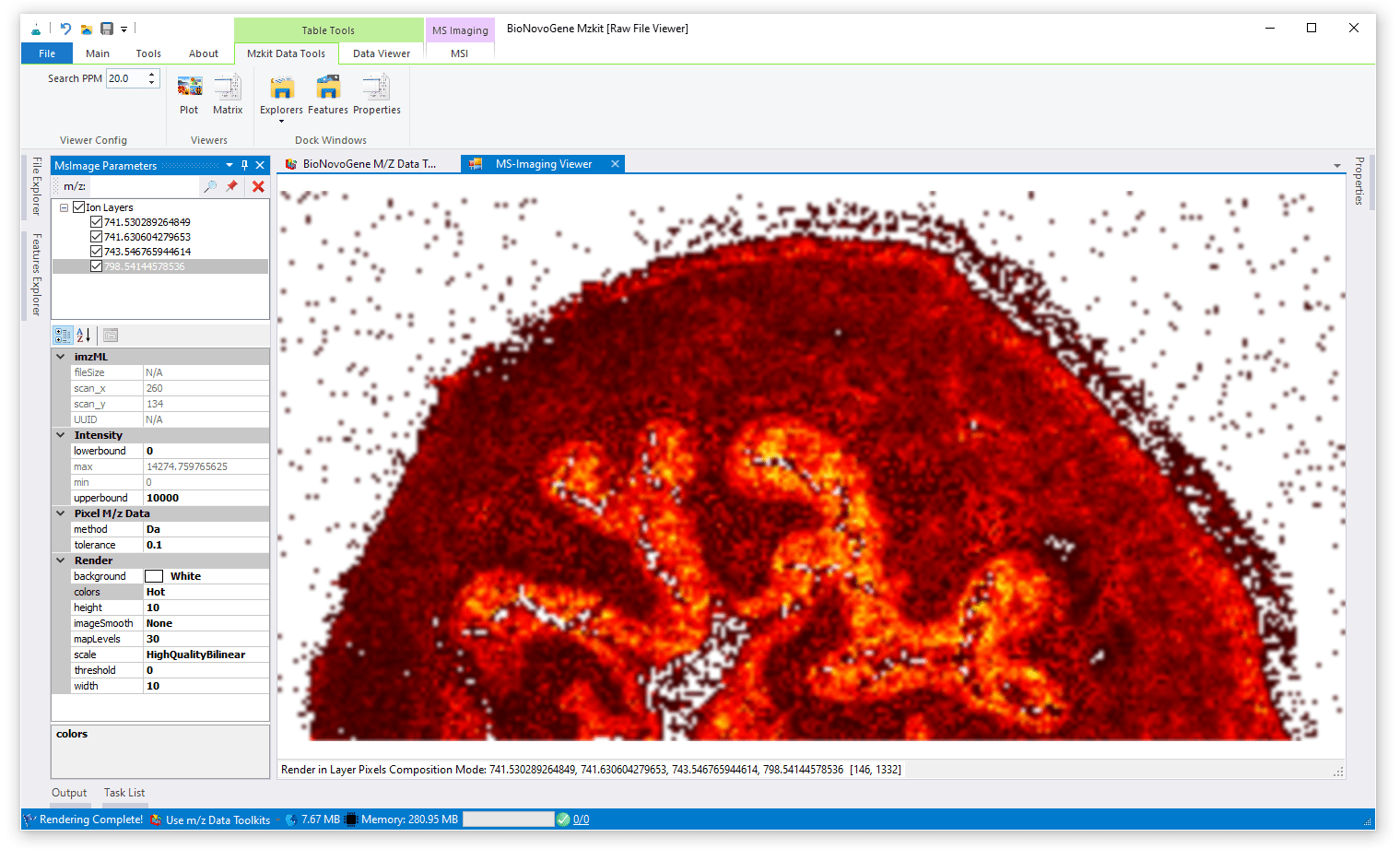
对于离子信号混合渲染模式,我们一般是采用Jet颜色谱或者Hot颜色谱来进行渲染。当然ColorBrewer颜色谱也是一种非常不错的渲染选择。
- 单细胞视角下的微生物基因组代谢酶嵌入分析 - 2026年2月25日
- 基因组代谢酶层级嵌入 - 2026年2月23日
- 基因组功能吉布斯LDA主题建模 - 2026年2月23日

17 Responses
Marvelous, what a weblog it is! This web site
provides helpful facts to us, keep it up.
Pretty! This has been an extremely wonderful article.
Thank you for providing this info.
Скажите, пожалуйста, есть ли учебные материалы по программному обеспечению mzkit, упомянутому в статье? Я искал в интернете, но не нашел много соответствующей информации.
you can try search video on bilibili.com
Очень интересно! Здесь предложен новый универсальный бинарный формат файлов для хранения данных масс-спектрометрии. Не могли бы вы подробно рассказать об деталях реализации формата mzPack, упомянутого в вашей статье? Я считаю, что можно активно продвигать применение этого формата в области масс-спектрометрического анализа
well, errrrrrr. Although the mzPack file format offers excellent compatibility with all mass spectrometry data analysis applications, from LC-MS, GC-MS, and MRM to mass spectrometry imaging, it has a major drawback. As a binary file format, its readability is a significant weakness compared to the widely-used mzML file, which is in an XML text format. This may make it unsuitable for widespread adoption.
Hello. And Bye.
Excellennt blog riht here! Also your website lotss
upp very fast! What weeb hoost aare yoou the use of?
Cann I get your assocciate lunk tto your host? I want mmy websitee loaded up aas
quicklky as yours lol
The other day, while I was at work, my sister stole my apple ipad
and tested to see if it can survive a 30 foot drop, just so she can be a youtube sensation. My iPad is now broken and she has 83 views.
I know this is entirely off topic but I had to
share it with someone!
“Her rigorous scientific spirit deserves recognition! Suggest she livestream ‘High-altitude Free-fall Physics’ next time—title it On Gravity and the Shattering of Internet Fame Dreams. I’ll sponsor iPad #2 on one condition: crowd-fund me a blast-proof glass cover first 😎”
Cool + for the post
I’ve been surfing on-line more than 3 hours nowadays, but I by no means discovered any fascinating article like yours. It is pretty value sufficient for me. Personally, if all webmasters and bloggers made excellent content as you did, the web will likely be a lot more useful than ever before.
Wow that was odd. I just wrote an extremely long
comment but after I clicked submit my comment didn’t appear.
Grrrr… well I’m not writing all that over again. Anyway, just wanted to
say superb blog!
Hi, thks for write your comment
[…] 在之前的博客文章之中,我们介绍了怎样通过使用mzkit的桌面端软件进行质谱成像的原始数据的查看操作:《【Mzkit教程】查看质谱成像原始文件》。通过基于图形界面的桌面应用程序,我们可以很方便的低门槛的进行原始数据查看。但是桌面端图形化界面存在一个缺点就是:图形界面的应用程序一般在个性化定制上存在难点,并且进行批量化的自动处理会比较困难。 […]
[…] 【Mzkit教程】查看质谱成像原始文件 […]
[…] 下面我就以最近编写好的一个在mzkit软件中处理质谱成像原始数据的计算任务,来向大家讲解我是如何在mzkit软件中进行应用程序管线模式的开发的。 […]