
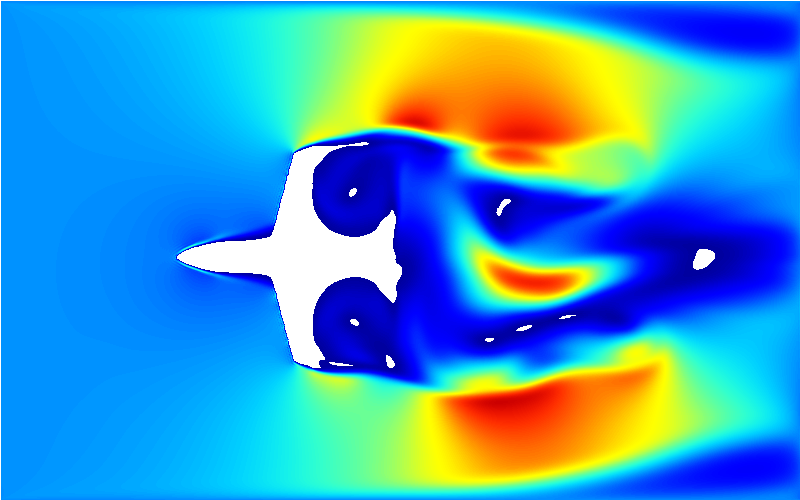
估计阅读时长: 30 分钟https://github.com/xieguigang/Moira LBM(格子玻尔兹曼方法)凭借其介观模型特性,在流体模拟领域展现出显著技术优势:其碰撞与迁移过程仅依赖局部数据,天然适配GPU并行计算,CUDA实现可达成10–100倍加速比;处理复杂几何边界时无需生成体网格,通过格点标记固体并配合反弹边界即可高效实现,尤其适用于多孔介质等场景;同时,通过扩展分布函数可灵活耦合多物理场,例如引入温度分布函数模拟传热,或采用伪势模型捕获多相流中的相分离现象。尽管在高速或高粘度流动中存在局限,但通过MRT算法优化及GPU硬件加速,LBM已成为微流动、多孔介质、多相流等复杂流体模拟的理想工具,在航空工程等领域已有成功应用案例,其应用前景持续拓展。 Order by Date Name Attachments frame-00093 • 2 MB • 871 click 2025年8月9日ffmpeg • […]
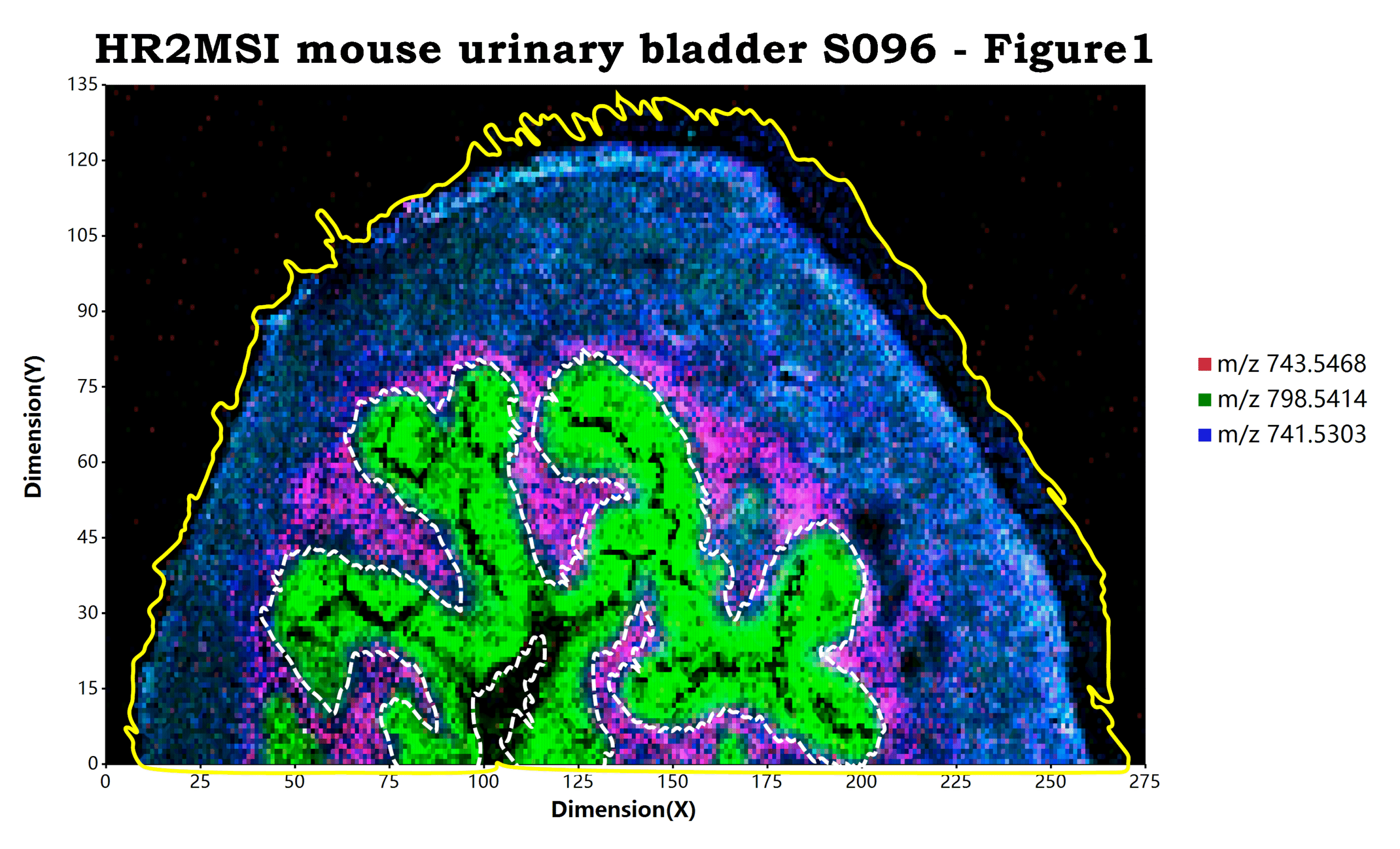
估计阅读时长: 10 分钟目前经过改进和优化之后的基于mzkit代码库底层的msimaging质谱成像软件包在样本可视化上进行了非常多的改进,诸如: 添加样本原始背景叠加 目前进行质谱成像可视化,程序包不仅仅可以使用任意rgb纯色来作为可视化的背景。目前还可以支持直接使用原始数据的背景作为质谱成像的显示背景。进行这个显示的秘诀就在于简单的在脚本中添加一个TIC背景图层:geom_MSIbackground("TIC") ggplot(msi_data, padding = "padding: 200px 600px 200px 250px;") + geom_MSIbackground("TIC") # rendering of […]

估计阅读时长: 12 分钟https://github.com/xieguigang/Microsoft.VisualBasic.Drawing 最近在Linux服务器上面搞数据分析,因为Linux服务器只能够是通过SSH远程登陆上去的,没有图形化界面,所以想查看生成的结果图的话,只能够将图片文件通过FileZilla工具从服务器上下载下来在本地查看。这种方法非常的繁琐,至少相对于在服务器上跑完了程序后直接查看结果这样子的操作要复杂一些。 如果要能够直接在Linux服务器上查看图片,可行的一个方法就是,如果你有服务器的Root权限的话,可以将你的目录通过smb协议共享出来,在windows上挂在为共享文件夹,这样子在Linux服务器上跑完命令后,再回到Windows的Explorer程序上刷新一下。但是这个对于网络地理位置较远的服务器而言,可能网络速度不是很好,对于几十兆的图片结果文件,可能刷新会存在延迟,你可能需要刷新好几次才会更新Windows上的图片缩略图;并且通过smb开放共享文件夹你还需要记住smb的第二套账号密码,如果账号密码过于简单,那么你的Linux服务器上的数据安全性就会存在问题。 另一个方案就是通过SSH-FS方案,通过你的ssh账号将远程Linux服务器挂载为本地硬盘,来查看服务器上生成的图片文件。但是这个也和上面的方案一样会受限于网络传输速度的影响。 看来,我们只能够在Linux的终端上想办法来进行图片文件的查看了。 Order by Date Name Attachments Capture • 269 kB • 882 […]
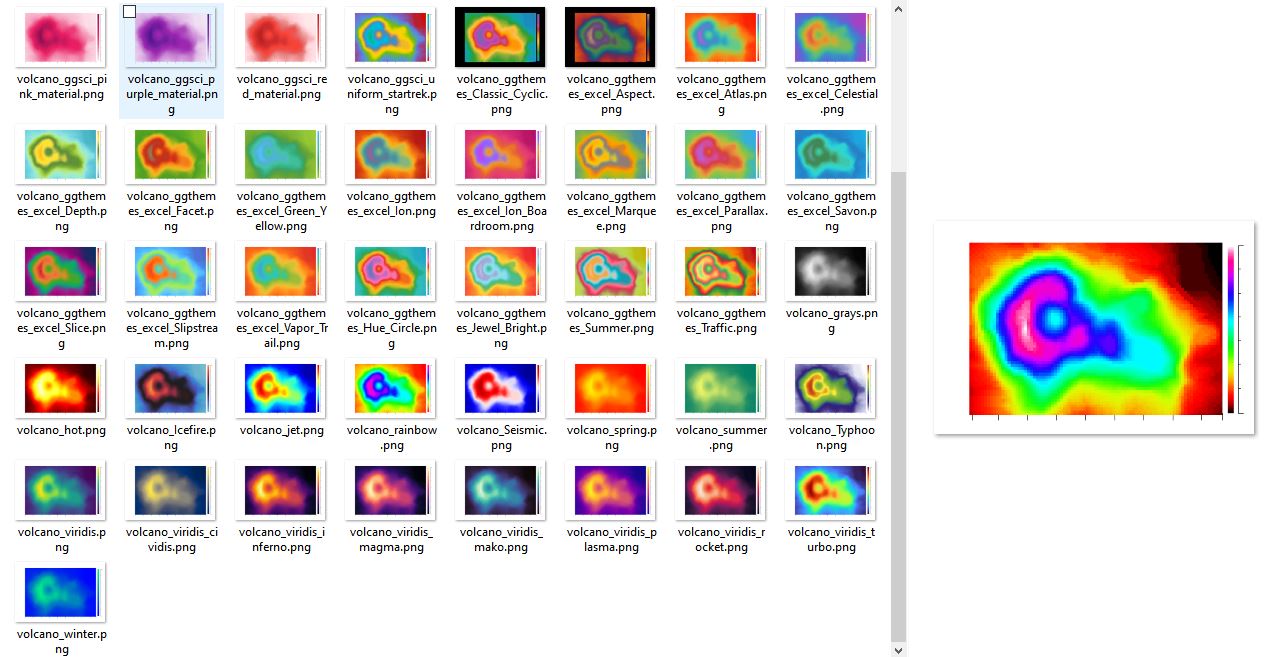
估计阅读时长: 11 分钟在进行热图的渲染的时候,我们需要首先将需要进行渲染的数据转换为一个0到1之间的灰度值,然后基于所设定的颜色列表,将灰度值映射为颜色列表的索引号,获取某一个灰度对应的颜色,从而完成对热图的渲染过程。在这个过程中,假若我们是针对热图需要获取得到一个连续的颜色列表,则我们还需要使用插值算法针对基础的关键颜色列表进行插值计算,生成调色板。 Order by Date Name Attachments volcano_ggthemes_Traffic • 17 kB • 756 click 2025年6月12日volcano_ggthemes_excel_Ion_Boardroom • 15 […]
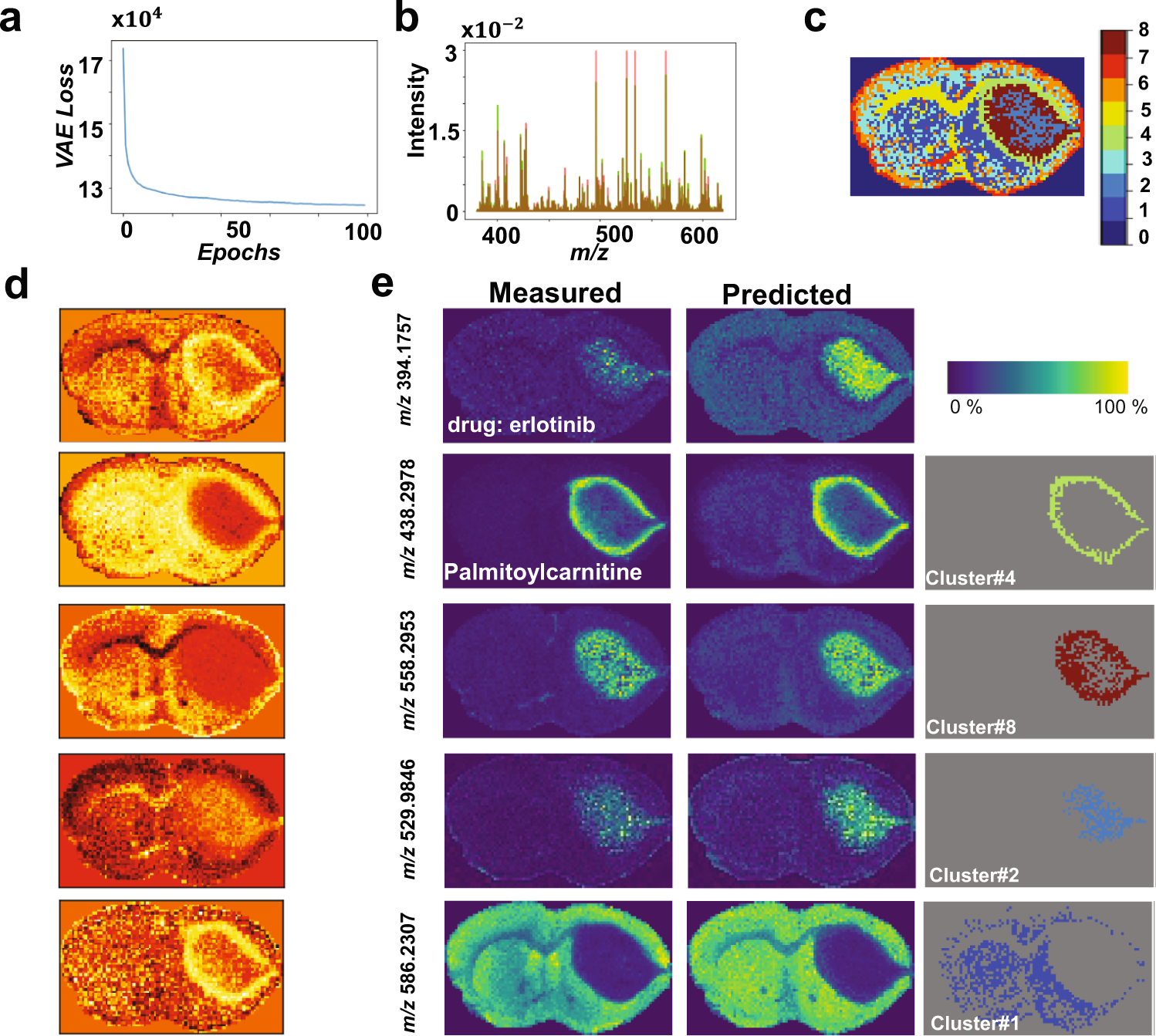
估计阅读时长: 4 分钟基于UMAP工具进行简单的自动化组织分区操作 在这里我们假设已经可以正常的将空间代谢数据导入至MZKit工作站软件之中。假若需要借助于MZKit工作站软件进行切片组织样本的自动化分区操作,相关的功能可以在【MSI Analysis】菜单栏中寻找到。在这里我们打开【Show Map Layer】按钮,选择【UMAP and clustering】功能。 基于降维的组织自动化分区原理 因为降维操作一般是一种特征提取操作,所以经过降维之后,在高维度空间上无法显现的特征,在低维度会呈现出来。在高维度空间散落的相近的数据点,在经过特征提取之后,低维度上会产生相似的特征信息,相互聚集在一簇。这样子我们就可以在低维度空间上通过一些聚类算法讲这些特征进行聚类,最后将聚类特征结果标记到各个散点上的对应的原始成像空间上,我们就可以看见组织分区的结果了。 Abdelmoula, W.M., Lopez, B.GC., Randall, E.C. et […]

估计阅读时长: 24 分钟假若现在有两条Fasta序列放在你面前,现在需要你进行这两条Fasta序列的相似度计算分析。如果对于我而言,大学刚毕业刚入门生物信息学的时候,可能只能够想到通过blast比对的方式进行序列相似性计算分析。基于blast比对方式可以找到生物学意义上的序列相似性结果,但是计算的效率会比较低。假设现在让你使用这些序列进行机器学习建模分析,或者基于传统数学意义上的基于相似度的无监督聚类分析的时候,面对这些长度上长短不一的生物序列数据,可能会比较蒙圈,因为传统的数学分析方法都要求我们分析的目标至少应该是等长的向量数据。 Order by Date Name Attachments Fasta-A • 544 kB • 934 click 2023年6月29日visualize • 45 […]
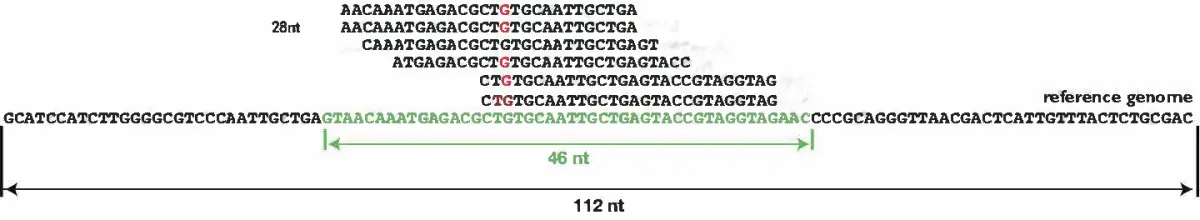
估计阅读时长: 11 分钟给定一组n个字符串数组,找到包含给定集合中每个字符串的最小字符串作为子字符串。我们可以假设这个字符串数组中没有字符串是另一个字符串的子字符串。那么基于上面的描述,我们就可以得到下面所示的问题求解目标: let arr[] = ["catg", "ctaagt", "gcta", "ttca", "atgcatc"] // output: gctaagttcatgcatc 上面的问题描述实际上是一个最短超字符串问题(shortest common superstring) Order […]

估计阅读时长: 5 分钟https://github.com/xieguigang/scale_colour_genshin 在用R绘图时,颜色设置是美化过程中不可缺少的一步。在实际绘图时,一般不会一一手动寻找合适的颜色,而是通过一些R包、网站提供好的,美观的颜色组合,即调色板(palette),可供使用。在这里介绍一种通过提取图片主题色的方法来为我们自动生成画图所用的颜色板数据。 Order by Date Name Attachments 383807b4 • 132 kB • 1020 click 2023年4月8日faruzan • […]
估计阅读时长: 8 分钟https://github.com/rsharp-lang/NRRD NRRD(Nearly Raw Raster Data)是一种用于存储类似于热图成像数据的文件格式。其实我们可以将NRRD看作为类似于bitmap之类的未压缩的原始光栅图像文件。只要我们有对应的解码方式,我们就可以像查看普通图片文件一样查看NRRD文件。 Order by Date Name Attachments raster__238 • 61 kB • 1037 […]
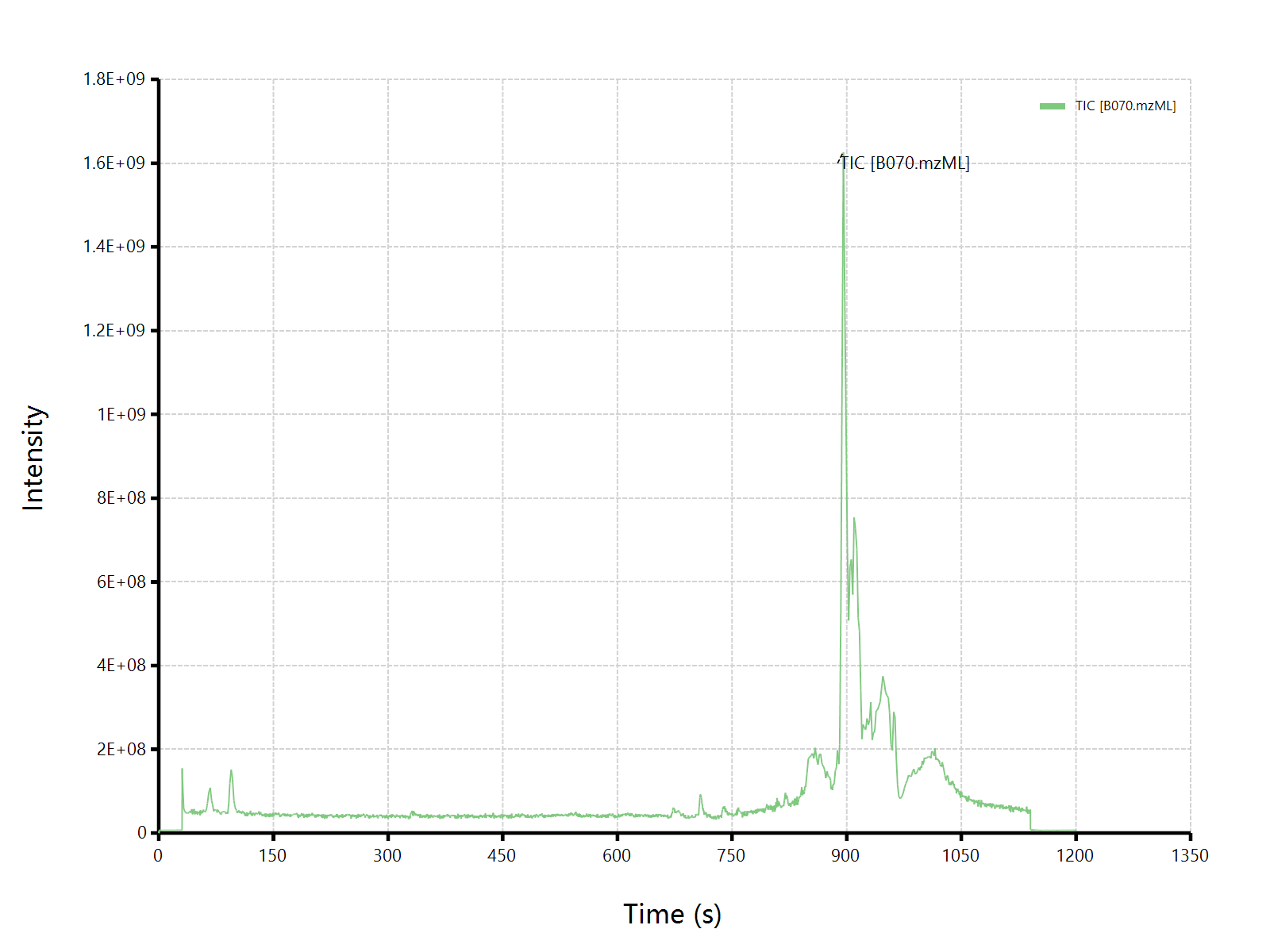
估计阅读时长: 5 分钟在BILIBILI上观看视频:《【BioNovoGene Mzkit教程】代谢组学原始数据处理基础》 最近我在B站的视频页面下发现了这样的一条评论,面对质谱数据分析领域内的初学者的求教,其实自己也是非常的诚惶诚恐的。因为在视频中所使用的脚本语言是自己开发的一门新语言,所以可能给一些初学者造成了一部分的困扰哈哈😅😄😅😅。首先先对这个粉丝说一声抱歉哈。 针对上述的提问,我的回答大概是有以下的几点: Order by Date Name Attachments question_20230223 • 17 kB • 879 click […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?