
文章阅读目录大纲
大家好呀,今天的这篇文章主要是为了回答在B站上的一位小伙伴的请求

这位小伙伴提到的处理ap-maldi的Github上的代码,大概就是指的是MZKit了。那今天在这里就来仔细讲讲MZKit工作站软件在质谱成像原始数据查看上的一些操作吧。
首先来给大家介绍一下MZKit软件:MZKit工作站软件是一款代谢组学原始数据分析软件,其目前集成了LCMS/GCMS/MALDI以及靶向定量原始数据的数据分析功能,除了质谱数据分析和查看,在MZKit中目前还集成有化学信息学以及生物信息学相关的功能模块。通过这些功能模块,可以实现将质谱原始数据和具体的生物学知识进行关联。在今天我们所介绍的质谱成像数据文件查看模块,其就属于MZKit工作站的一个功能模块。如果想要访问质谱成像功能模块,我们可以通过软件主界面的【MALDI Viewer】菜单进入。

软件主界面介绍
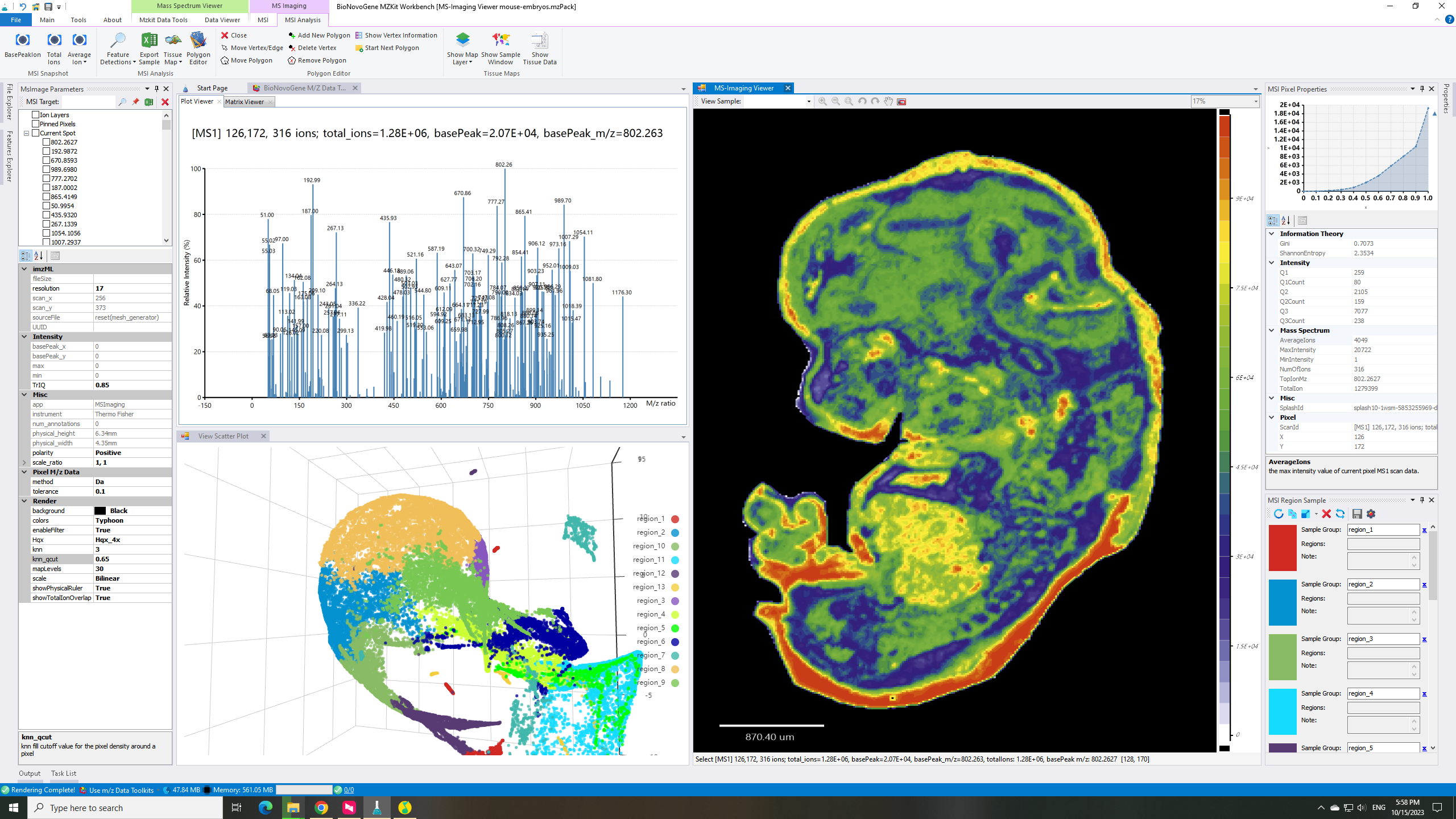
在打开了质谱成像功能模块之后,我们可以看见软件有如下界面模块所构成:
![]()
-
- 顶部菜单栏,主要有两大部分组成:【MSI】和【MSI Analysis】,其中第一组【MSI】菜单主要是应用于质谱成像原始数据文件的处理操作,例如:文件导入操作,文件导出操作,文件合并操作,文件数据旋转操作,质谱成像效果调整等操作;而第二组菜单主要是应用于质谱成像数据的一些简单分析处理操作,例如:获取文件中的离子特征列表,样本手动分区,导入和导出样本分区文件等基础的数据操作功能。
-
- 左侧的质谱成像参数调整窗口【MSImaging Parameters】,在这个窗口中又分为上下两部分:其中窗口的上部分为离子列表区域,在这个离子列表区域中,可以通过手动输入离子或者从表格中选取离子列表来进行目标离子的成像可视化;窗口的下半部分则是质谱成像的参数调整部分,具体的参数内容可以详细的查看本文后续的内容
-
- 软件中间占据很大一块面积的黑色区域就是质谱成像的显示区域了,后续质谱成像的显示都是在这个区域内展示的。在这个区域内同时又可以分为左右两部分:左边的黑色部分就是质谱成像显示的画布区域,右边则是成像热图的颜色条和具体的离子响应强度值
-
- 软件右边部分则是质谱图属性窗口。在这里会展示我们在质谱成像显示画布上通过鼠标点击,选取的某一个坐标斑点对应的质谱图的一些简单统计信息,例如:信息熵,基尼系数,最大响应度以及对应的离子mz,离子峰数量,质谱图的Splash-id等
原始数据文件打开
在这里我们主要介绍MZKit软件常用的几种空间代谢原始数据文件的打开操作。在MZKit工作站软件中打开原始数据文件是一件非常简单容易的事情,我们只需要在【MSI】菜单组之中,找到最左边的【Open】打开按钮,使用这个打开按钮就可以非常简单的进行空间代谢原始数据文件的打开操作了。现在我们点击这个菜单按钮,在弹出来的文件打开对话框中,可以看见目前在MZKit中支持有如下文件格式:

*.raw*.udp*.imzML*.mzPack
对于上面所列举出来的常见的文件格式,我们只需要直接打开即可,除了直接打开赛默飞的RAW文件需要输入扫描的目标样本的长和宽信息,打开其他的文件格式不需要输入额外的信息。
特别说明1:打开赛默飞RAW文件
对于打开使用传统的LCMS文件格式所储存的空间代谢原始数据,例如赛默飞RAW文件,mzML或者mzXML文件。因为在里面是没有任何空间坐标信息的,所以对于打开这类原始数据文件,我们需要输入一些额外的信息来帮助MZKit工作站软件进行坐标点信息的计算操作。例如在我们打开上述的没有包含有空间坐标信息的原始数据文件之后,MZKit软件会发现没有对应的坐标信息,会弹出一个输入窗口,让我们输入对应的切片样本的扫描的宽度和高度信息:

在这里我们根据实验室的上机人员所提供的扫描软件的参数信息,输入正确的宽度和高度信息,然后点击【OK】按钮就可以打开我们的原始数据文件进行质谱成像可视化了。
特别说明2:导入10x genomics空间转录原始数据
在MZKit软件之中,你还可以将10x genomics的空间转录原始数据进行导入,可视化生物样本上的基因的表达热图。具体的操作为:在【MSI】菜单栏中找到【Imports 10x Genomics】按钮,在弹出来的对话框中,一般会分为下面的两种情况进行相应的文件输入操作:

- 已经完全处理好的10x Visium原始数据文件,h5ad原始数据文件里面应该包含有空间坐标信息,表达矩阵,基因注释信息,组织分区信息等,这个时候可以仅输入目标h5ad原始数据文件即可
- 对于通过Space Ranger初步处理的空间转录组原始数据文件,除了输入h5原始数据矩阵文件外,还需要输入Space Ranger软件输出的基因列表,barcode信息文件和扫描坐标信息,MZKit软件可以将这些信息进行组装,合成一个完整的空间转录组输入进行后续的查看操作。
查看质谱成像热图
对于质谱成像可视化而言,其就是一个热图的绘制过程:我们将一定的离子图层信息提取出来,根据所提取的离子图层的信号响应度信息进行线性变换,映射到对应的颜色上,然后吧颜色显示到对应的空间坐标位置,就可以通过热图的方式吧我们的生物样本可视化出来了。在MZKit工作站软件之中,可以非常简单的进行单个离子的质谱成像,以及多个离子的质谱成像查看。
1. 单个离子质谱成像
如果需要进行单个离子的质谱成像,我们只需要在【MSImaging Parameters】窗口的输入栏中填入我们的目标离子的m/z值,然后点击输入框旁边的放大镜按钮即可查看目标的质谱成像图。例如我们想要查看m/z值为643.0663
- 找到位于软件界面左侧的【MSImaging Parameters】窗口,
- 在顶端的【MSI Target】输入栏中输入目标离子的m/z值643.0663,
- 最后点击输入栏右边的放大镜按钮即可进行成像图的查看
注意,有些时候假若侧边栏的宽度不够,可能输入框旁边的按钮会被自动隐藏起来,这个时候你可以通过点击输入框旁边的小三角形将这些自动隐藏的按钮显示出来;当然,也可以通过拖拽窗口的右边拉宽窗口的宽度,当宽度足够的时候,输入栏右侧的按钮会全部显示出来。

特别说明:通过化学式获取m/z值
通过m/z值进行质谱成像图的查看,这个需要老师您明确的知道目标代谢物的精确分子质量和质谱母离子加合物信息进行计算。但是假若老师您并不清楚这个计算过程,也可以通过输入化学式的方式来进行成像,例如在这里我们想要了解Adenosine-5'-triphosphate
- 我们可以通过目标代谢物的名称在代谢物数据库中进行搜索,例如通过NCBI的PubChem数据库进行搜索操作,得到ATP目标代谢物的化学式信息为:C10H16N5O13P3
- 将我们获取得到的化学式信息复制到【MSImaging Parameters】窗口中的输入栏
- 然后同样也是通过点击输入栏旁边的放大镜按钮进行计算
- 之后,MZKit工作站软件会根据所输入的化学式信息计算出精确分子质量,然后再根据不同的母离子加合物类型计算出目标mz值,并列举在【Ion Layers】列表之中
- 最后,选择一个计算出来的m/z值,通过鼠标右键快捷菜单中的【Render】菜单进行成像操作

通过PubChem数据库搜索代谢物的名称,然后获取得到【Molecular Formula】信息

特别说明:通过集成的Pubchem查询功能获取成像离子列表
除了可以直接在【MsImaging Parameters】输入框中输入化学式进行mz值列表的计算,老师您还可以更加方便的使用MZKit集成的PubChem数据库查询功能进行成像离子列表的计算获取。具体的操作如下:
- 在【MSI】菜单栏中,点击【Search PubChem】菜单按钮
- 接着在弹出来的对话框中输入需要进行查询的代谢物名称,例如在这里我们搜索
Adenosine-5'-triphosphate - 接着就可以点击输入框右边的【Search PubChem】按钮
- 等待一会,对话框中会显示进行名称匹配的化合物候选列表,里面应该是包含有具体的名字,分子式,精确分子质量,CAS编号信息
- 这个时候,我们选择某一个希望需要进行显示的目标化合物
- 可以根据需要调整mz的匹配误差为ppm误差或者分子质量差绝对误差
- 点击【OK】按钮进行计算

- 最后我们可以通过表头选中某一个离子的行数据,通过右键快捷菜单的【View】操作查看目标离子mz的质谱成像图

在上面搜索出来的表格中,老师可以简单的通过【pixels】列中的数值来判断对应的mz是否存在于样本切片上:假若pixels值等于零,则是不存在该mz,任意大于零的pixels数值意味着老师可以在切片上找到该离子mz的成像图
2. 三个离子叠加质谱成像
上述的单个离子成像,可以仅能够可视化单个代谢物离子在我们的目标生物样本切片上的分布情况。如果我们想要查看多个代谢物离子在样本上的分布情况的话,这个时候我们可以通过RGB三离子叠加质谱成像来完成。如果要进行三离子叠加成像,我们可以通过下面的步骤来完成:
- 首先我们需要准备需要进行可视化的三个目标离子的mz值,然后通过【MSImaging Parameters】窗口中的输入框输入MZKit
- 在这里,我们不使用放大镜按钮进行查看,而是通过放大镜按钮旁边的图钉按钮将我们的目标离子添加进行候选列表之中
- 接着在候选离子列表之中,勾选中需要进行成像的离子列表
- 通过鼠标右键快捷菜单,选择【Render(Layer Composition Mode)】功能菜单
- 接着在弹出来的对话框之中,设定好目标离子分别对RGB颜色通道的映射关系
- 点击【OK】按钮执行可视化操作,之后就可以看见三个代谢物离子在我们的样本上的分布情况了



在RGB模式下的三离子叠加成像图中,不同的像素点的颜色来自于RGB三原色通道中对应的离子响应强度的线性变换的组合结果。在此模式下,会存在有如下所示的分布结果:
- a. 假若某一个区域呈现红色,蓝色或者绿色的任意一种颜色,则表示该区域内颜色对应的代谢物离子是单独分布在该区域的
- b. 假若某一区域呈现除了红绿蓝三原色以外的其他颜色,则表示该区域存在有两种或者三种代谢物的分布情况
- c. 假若某一区域内是呈现黑色的情况,则表示该区域内没有成像的三种代谢物存在于该位置
3. 特别说明:空间转录组成像可视化
MZKit工作站软件除了可以进行基于质谱数据的空间代谢原始数据成像操作,还可以进行10x Genomics空间转录组的数据成像可视化。我们在这里假设老师您已经可以成功的通过MZKit软件相关的功能将10x的h5ad原始数据文件导入到MZkit软件之中,则后续的在MZkit软件之中的基因空间表达成像操作和代谢物的空间表达成像操作完全一致;稍有不同的是,成像的目标数据来源会和空间代谢组的操作存在有差异:对于空间转录组的成像,我们需要使用h5ad内部的基因注释结果来完成,加载基因注释结果,可以通过【Load All Annotation Layers】操作来完成:

- 在完成了10x的h5ad原始数据文件导入操作之后,在【MSI Parameters】窗口之中,通过鼠标右键菜单【Load All Annotation Layers】操作加载基因注释列表
- 完成加载操作之后,可以发现:在【Ion Layers】列表中会出现我们的原始数据文件中的基因列表
- 选择任意一个基因id,按照单离子成像操作进行【Render】操作,即可成像单个基因在样本上的空间表达分布情况
空间成像效果参数调整
进行空间成像效果参数的调整,可以让我们针对成像图进行个性化调整,目前在MZkit工作站软件之中进行成像参数的调整,可以通过【MSImaging Parameter】窗口来完成:
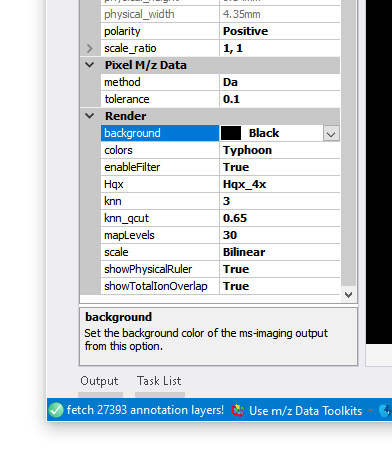
首先,我们在【MSImaging Parameter】窗口中找到【Render】参数调整区域,在这里面一般可以通过下面的参数来个性化调整成像图:
- 【background】这个参数可以修改画布的背景色,默认背景色为黑色
- 【colors】这个参数可以调整进行质谱成像图渲染所使用的颜色列表,这里一般推荐MZKit软件默认的viridis,其次是Typhoon,turbo,FlexImaging
- 【enableFilter】这个参数可以设定渲染器是否应用滤镜进行成像热图的优化,某些特定情况下,假若成像效果非常差,可以通过打开或者关闭这个选项来获取更好的成像效果
- 【Hqx】这个参数可以设定成像图的放大等级,这个参数的选项有None,不进行放大,以及2x,3x,和4x倍数的放大操作
- 【mapLevels】这个参数可以设定成像图所使用的颜色列表的插值等级,插值等级越高,色颜色越丰富,显示的图像可能会越细腻,这个参数的取值范围在10到255之间
- 【showPhysicalRuler】这个参数可以调整是否显示比例标尺
- 【showTotalIonOverlap】这个参数可以开关是否将整个切片的总离子成像热图叠加在我们的成像图的底层。打开这个选项在单离子成像图渲染的时候,显示出我们的样本切片的轮廓信息。
特别说明:调整成像图渲染滤镜
通过调整成像图的渲染滤镜,有些时候可以让我们获取得到更好的成像画面效果。我们可以通过顶端的【Tweaks MSI Filters】菜单打开滤镜的编辑器:

a. 成像图滤镜需要在【Render】参数中将【enableFilter】选项设置为True才会生效
b. 滤镜的不同生效顺序会产生不同的优化效果,可以通过鼠标右键菜单在【Raster Filters Pipeline】列表框中将对应的滤镜选项【上移】或者【下移】来调整滤镜模块的生效顺序,越靠前的位置,会首先生效
c. 可以通过滤镜选项前面的复选框是否处于选中状态来开关对应的滤镜模块,仅有处于被选中状态的滤镜模块才会生效
下面具体说明一下滤镜的具体实现功能:
- denoise滤镜可以调整噪声点过滤的功能,假若我们的成像图上具有很多单独离散分布的噪声点,可以通过这个模块在成像的时候过滤掉这些噪声点
- log滤镜可以针对成像的信号相应强度值取对应的log值,减少最大值和最小值之间的差距,凸显出更多的成像图细节
- TrIQ滤镜模块的功能和log滤镜的功能相似,也是用于减少最大值和最小值之间的差距,凸显出更多的成像图细节。一般情况下TrIQ的处理效果要优于log滤镜
- knn_fill滤镜模块一般是针对于我们不希望进行噪声过滤,而是希望将这些噪声点给更清晰的可视化出来的情况下使用。这个滤镜模块会使用knn方法对噪声分布的区域进行插值计算,使噪声区域的成像效果更明显
- soften滤镜模块通过卷积插值计算,将成像图进行柔化操作,可以让我们的成像图更加的光滑
导出空间成像图片文件
那,前面我们很详细的介绍了成像图的生成以及相应的个性化调整操作,那如果我们需要将调整好的成像图保存为文件呢?这个时候我们可以通过显示空间成像热图的画布上的右键功能菜单来完成了。一般而言,我们可以通过下列方式导出我们的成像图结果:

- 【Save Image】可以直接将目前画布上的图片结果保存为文件,这种方式导出的文件是带透明底的png文件,可以供编写文章的时候在Photoshop或者PPT中与其他的图标进行拼接生成插图的时候使用
- 【Copy】可以直接将目前画布上的成像图结果复制到剪切板上
- 【Export Plot】则是将带有坐标轴信息,以及颜色标尺信息的成像图导出为文件
导出成像图文件
下面我们重点介绍下通过【Export Plot】菜单将成像图导出为文件的操作。通过这个菜单操作,我们可以导出单离子成像图和RGB三离子叠加成像图为文件。在完成了相应的离子的成像热图渲染,以及个性化调整之后,我们可以通过这个右键菜单进行文件导出操作。点击这个操作菜单,MZkit软件会首先弹出一个设置对话框,如下所示:

由于成像的目标离子对象,颜色,滤镜等参数我们在导出操作之前已经完成了调整了,所以在这里我们仅只需要输入文件的保存路径以及图片的尺寸等信息即可:
- 【Image Save File】在这个输入框中输入文件的保存路径,可以通过点击右边的【...】按钮弹出的文件对话框来快捷的设定文件输出路径
- 【Size】在这里设定图片文件的大小尺寸
- 【PPI】在这里设定图片绘制的时候的dpi信息,一般而言,当图片的输出【Size】越大的时候,对应的PPI值应该越高,此时图片上的文字才会越清晰
- 【Padding】参数可以控制成像图绘图区域的大小

组织空间分型结果查看
除了能够查看空间代谢的质谱成像图以外,在MZkit工作站软件中还可以查看老师的切片样本的组织空间分型结果。如果老师拥有诺米代谢的空间代谢分析报告结果,一般可以从结果文件夹中找到一个名称为TissueMorphology.cdf

- 切换到【MSI Analysis】菜单栏
- 找到【Tissue Map】下拉按钮
- 点击【Imports Tissue Morphology】菜单
- 在弹出来的文件对话框中,选择分析报告文件夹中的
TissueMorphology.cdf - 打开后即可看见覆盖在成像图上面的组织空间分型结果图

可以看见,在【MSI Analysis】菜单栏中右边的【Show Map Layer】按钮已经处于选中状态,这个时候会在成像图上叠加上空间分型图的结果。反之,如果想隐藏掉这个组织空间分型图,可以点击一次【Show Map Layer】按钮,取消其选则状态即可。
如果想要修改查看的空间分型图的透明度或者绘制的斑点大小,可以点击【MSI Region Sample】窗口中的【Config Tissue Map Layer】按钮,打开配置对话框。在对话框中修改【Opecity】或者【Spot Size】参数值即可。


特别说明:查看UMAP散点图
在交付的结果报告中的TissueMorphology.cdf


- 【MZKit】简单自动化组织分区 - 2023年11月5日
- 【MZKit教程】质谱成像原始数据文件查看 - 2023年6月29日
- 生物序列图嵌入算法 - 2023年6月29日
5 Responses
10X Visium结果导入之后,是可以跟代谢数据一起整合的吗?还是说,只是一个接口呢?
目前软件上提供了用于手动关联空间转录组数据和空间代谢组数据的功能,使用具体的功能可以通过成像画布上通过右键菜单依次打开【Samples】->【Add Spatial Tile】加载Space Ranger的空间斑点结果:在软件上进行手动旋转调整位置完成两个空间组学之间的空间位置关联,然后老师这边拿到空间位置关联信息后,就可以按照斑点的空间位置对应关系导出表达数据做关联分析了
空间Spot结果在下载到的mzkit文件夹中有示例吗?我试了试,不是10X结果中的tissue_positions_list.csv(软件选择此文件,直接error);在默认结果中也没找到类似的文件;
老师提到的这个问题,我这几天会写一个使用教程,老师后面可以用教程中对应的demo示例来了解下功能的使用
waiting and waiting.