
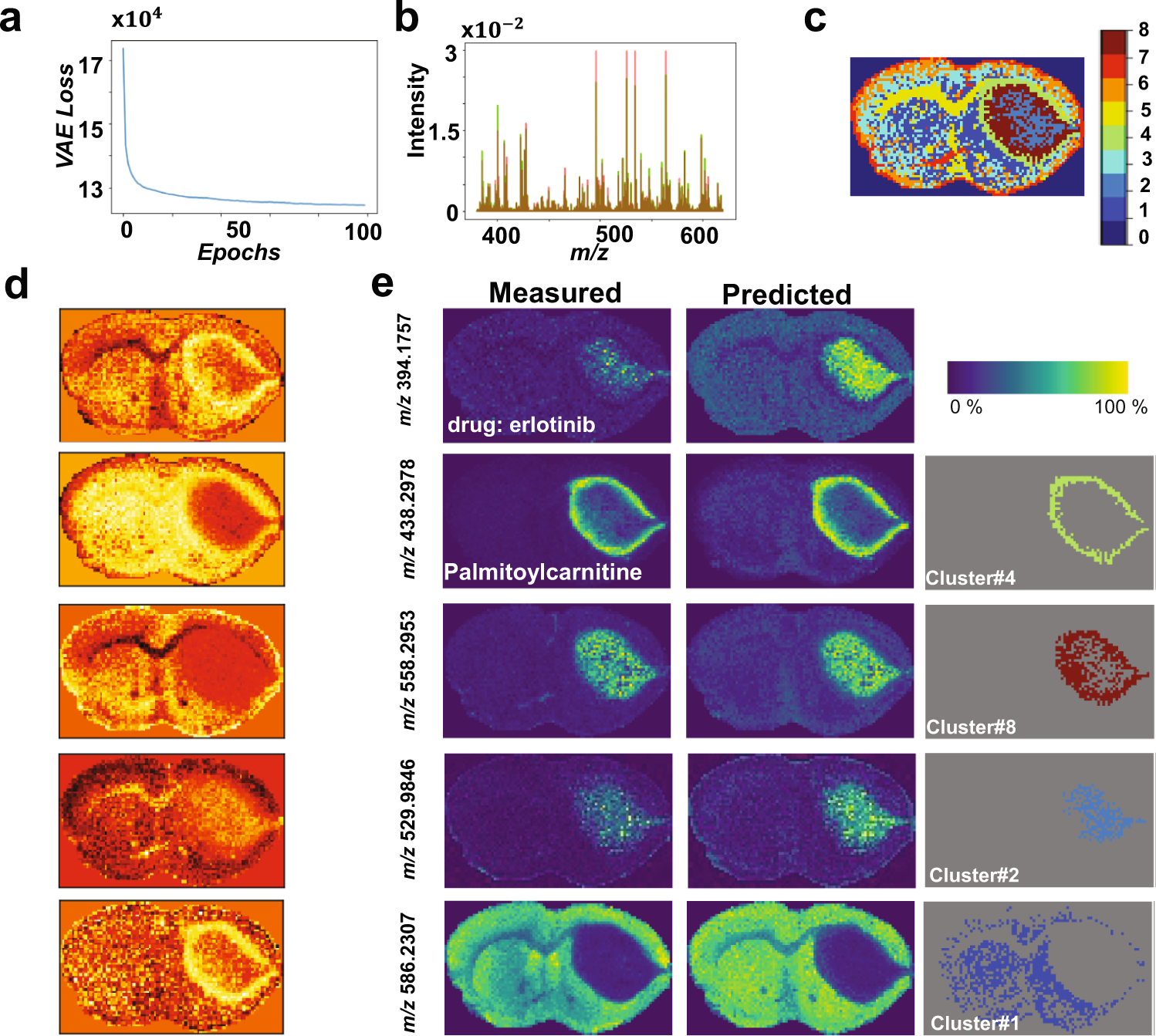
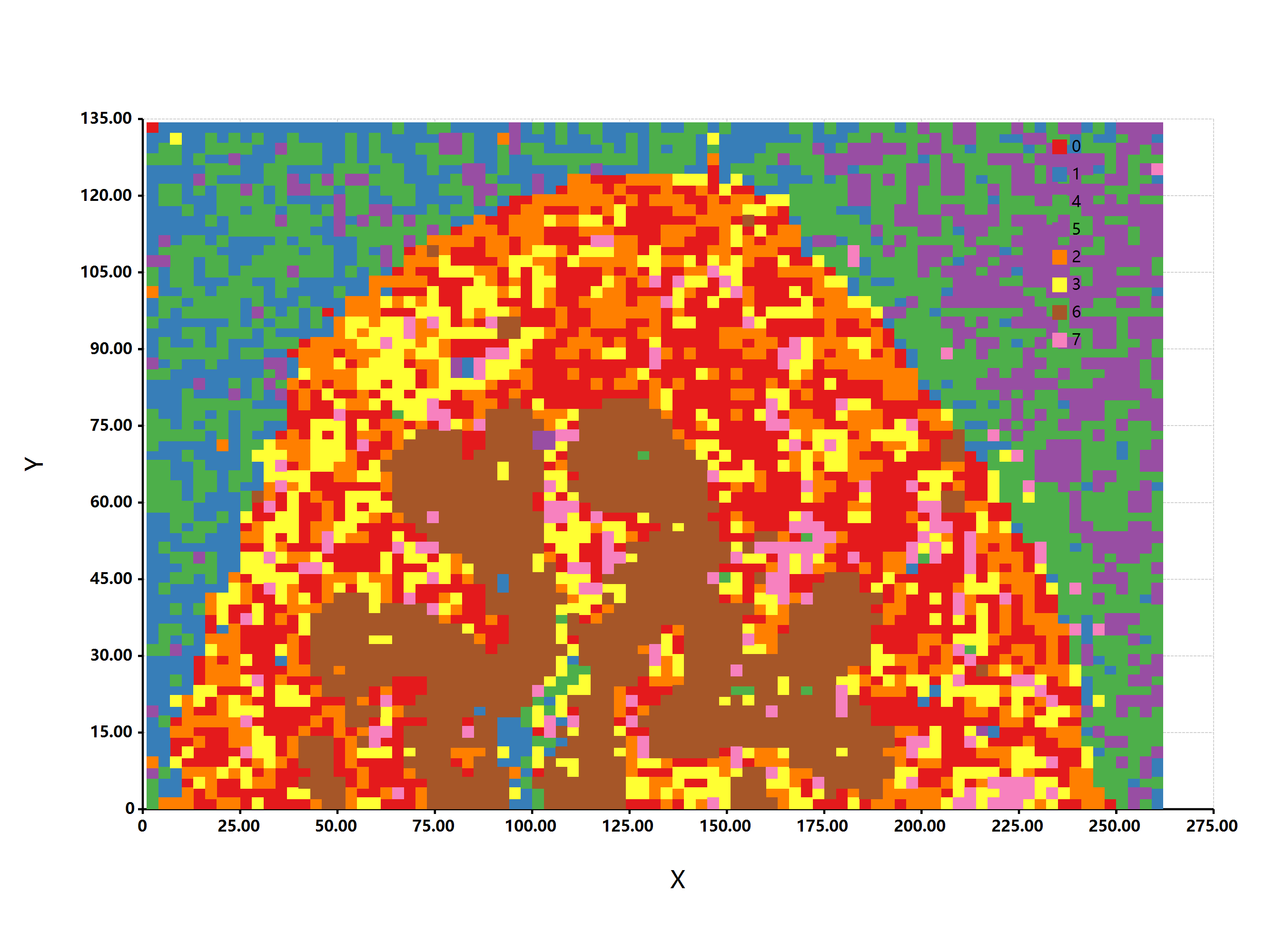
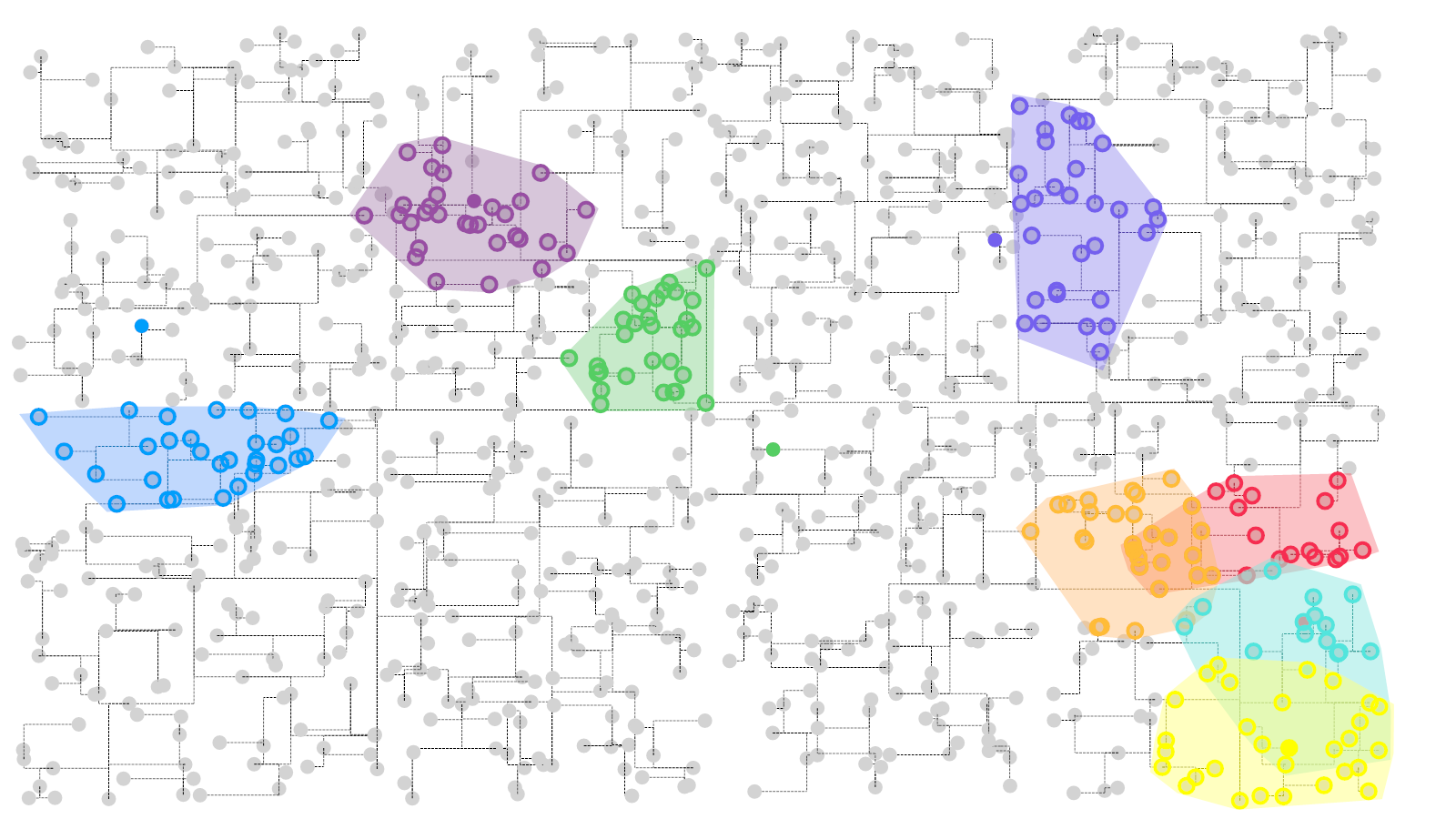
估计阅读时长: 4 分钟基于UMAP工具进行简单的自动化组织分区操作 在这里我们假设已经可以正常的将空间代谢数据导入至MZKit工作站软件之中。假若需要借助于MZKit工作站软件进行切片组织样本的自动化分区操作,相关的功能可以在【MSI Analysis】菜单栏中寻找到。在这里我们打开【Show Map Layer】按钮,选择【UMAP and clustering】功能。 基于降维的组织自动化分区原理 因为降维操作一般是一种特征提取操作,所以经过降维之后,在高维度空间上无法显现的特征,在低维度会呈现出来。在高维度空间散落的相近的数据点,在经过特征提取之后,低维度上会产生相似的特征信息,相互聚集在一簇。这样子我们就可以在低维度空间上通过一些聚类算法讲这些特征进行聚类,最后将聚类特征结果标记到各个散点上的对应的原始成像空间上,我们就可以看见组织分区的结果了。 Abdelmoula, W.M., Lopez, B.GC., Randall, E.C. et […]
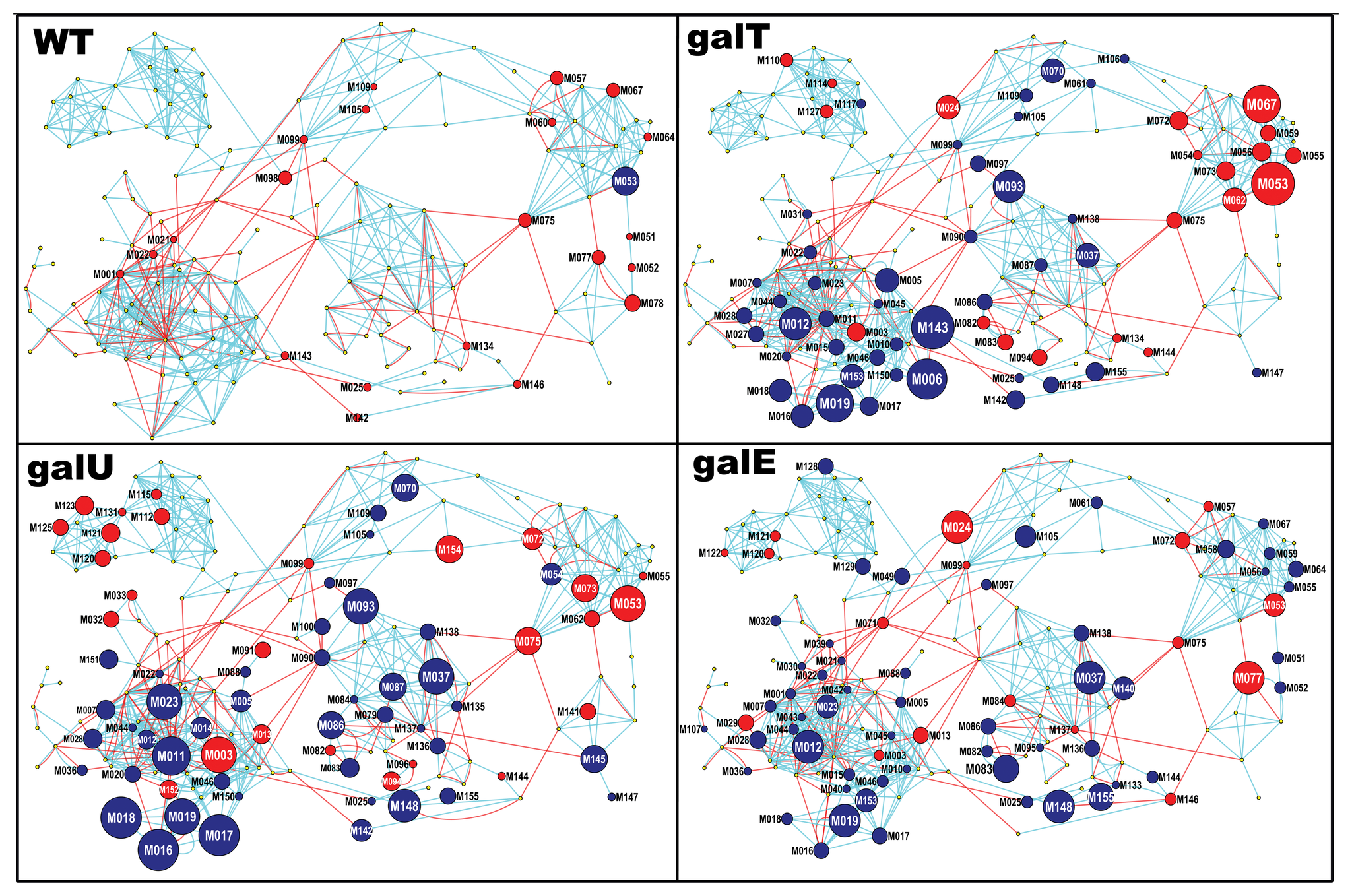
估计阅读时长: 5 分钟在工作之中可能会遇到需要进行两个网络图对象之间的相似度计算的情形:例如在质谱数据分析的化学信息学计算工作之中,我们在解析SMILES字符串得到分子图之后,可以基于图相似度比较计算方法来比较计算两个代谢物分子图之间的结构上的相似度。 Attachments pone.0078360.g003 • 2 MB • 856 click 2022年8月6日https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0078360
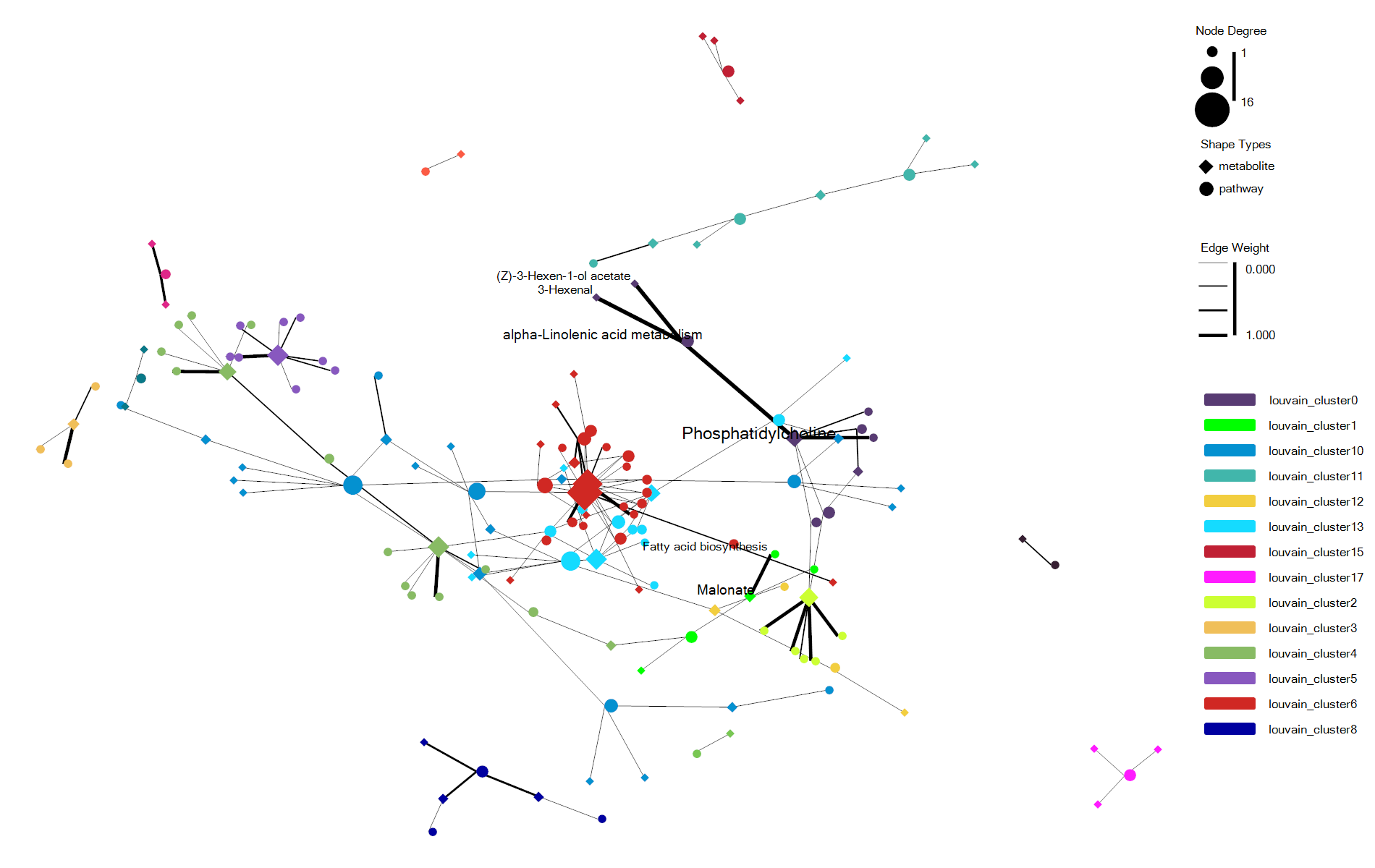
估计阅读时长: 7 分钟https://github.com/rsharp-lang/ggplot 在进行复杂关系的数据集进行可视化的时候,通过网络图的方式进行数据可视化可以让我们非常直观的借助于网络节点的聚集程度之类的布局信息了解到我们的复杂数据的关系结构信息。最近将R#语言之中的ggplot包进行网络可视化的代码库进行了一些更新。基于此功能更新工作,目前在ggplot程序包之中成功集成了ggraph程序包类似的网络可视化功能。在这里做了一些总结分享给大家。 Order by Date Name Attachments enrichNetwork_ggraph • 70 kB • 778 click 2022年6月1日enrichNetwork_ggraph2 • […]
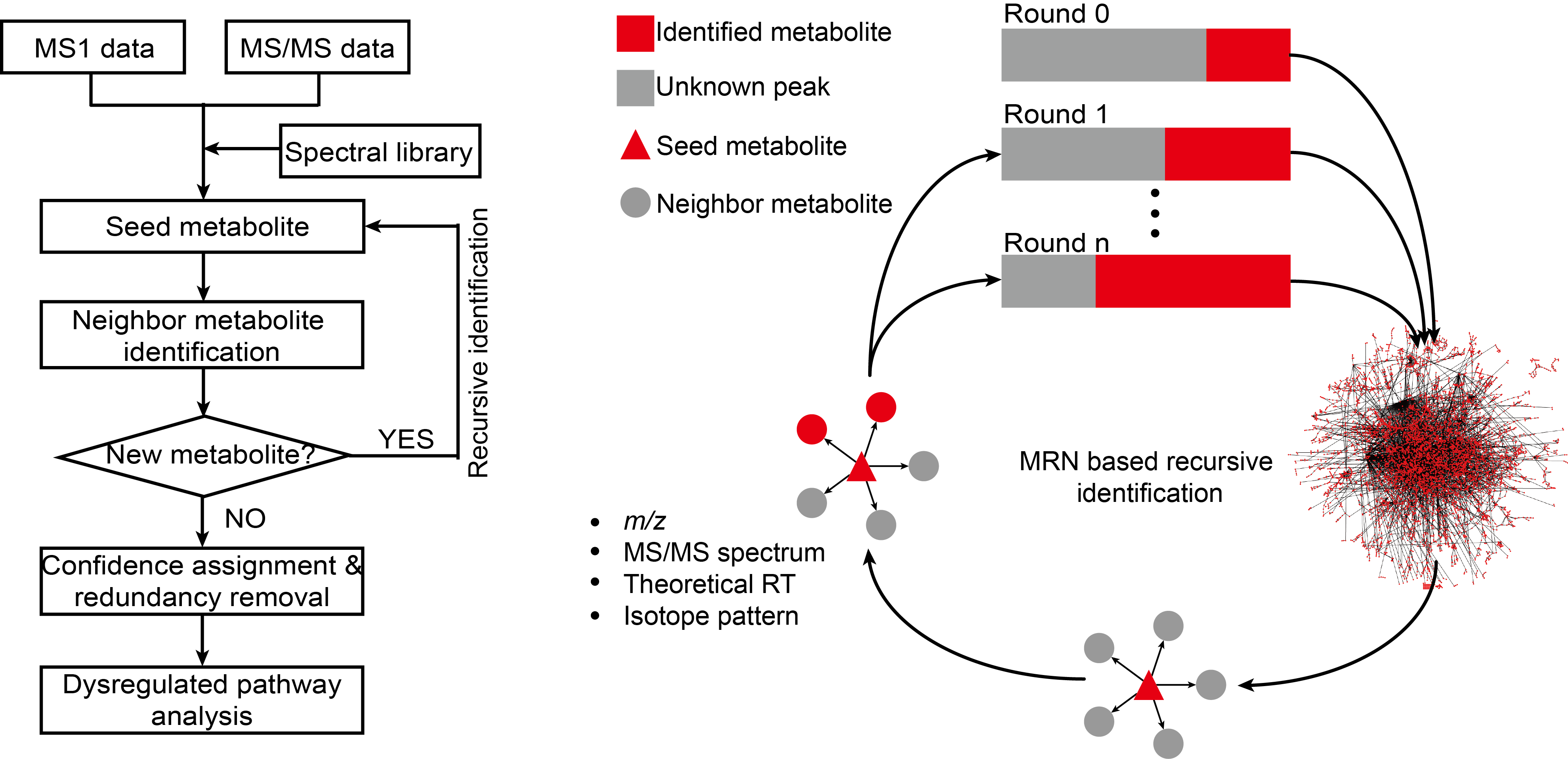
估计阅读时长: 6 分钟访问在线服务: http://metdna.zhulab.cn/ Metabolite identification is the long-standing challenge for liquid chromatography-mass spectrometry (LC-MS)-based untargeted metabolomics. Here, […]
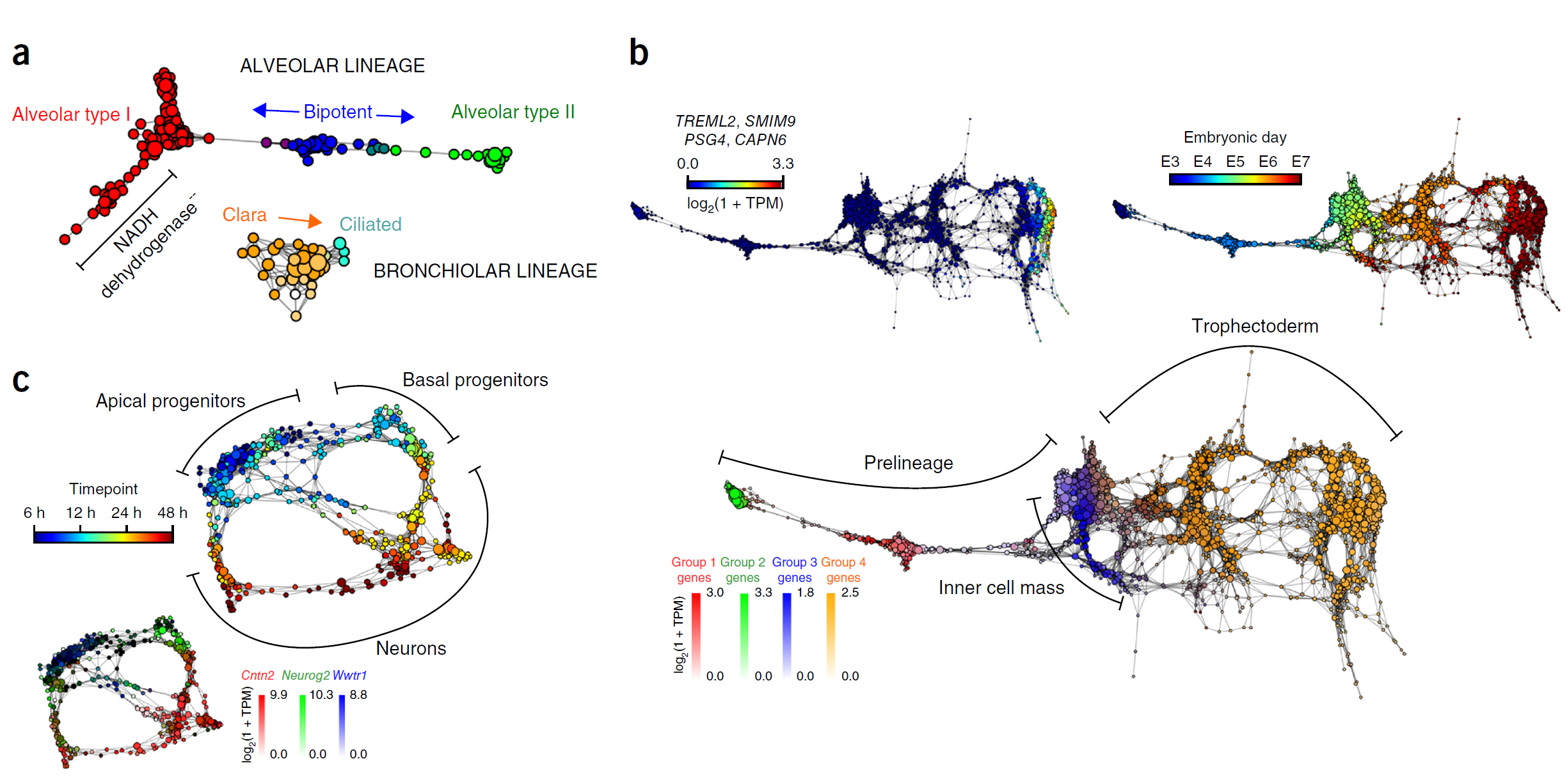
估计阅读时长: 14 分钟单细胞分析方法学习文献打卡记录: 【单细胞组学】PhenoGraph单细胞分型 【单细胞分析方法】VeTra:基于RNA速度的轨迹推断工具 【单细胞分析方法】单细胞图嵌入 Order by Date Name Attachments Cellular populations during motor neuron differentiation • […]
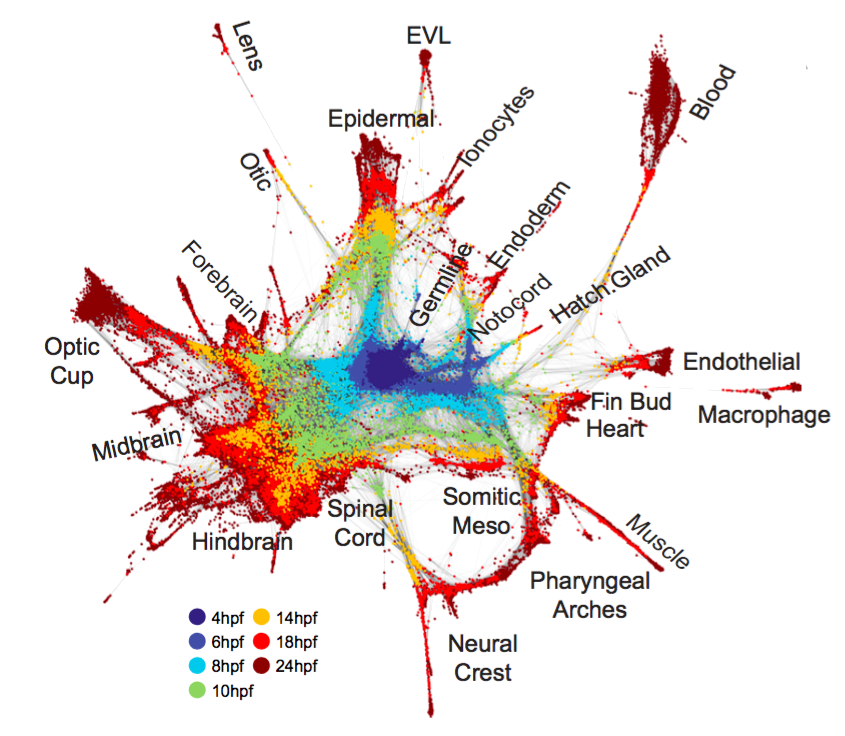
估计阅读时长: 7 分钟Assembles a manifold that is defined through a series of overlapping, locally-defined PCA subspaces. Non-mutual k-nearest-neighborhoods […]

估计阅读时长: 5 分钟https://github.com/xieguigang/graphQL 构建一个图数据库,可以用来帮我们解决复杂的知识关联计算问题。例如我们想要程序向我们回答dihydrogen oxide与water是否是同一个东西。如果光从字符串比较角度上面来看待这个问题的话,很显然,二者的字符串比较结果肯定是False。面对上面的这个问题,图数据库则可以很简单的向我们回答道上面的两个字符串都是指代的同一个东西。 Order by Date Name Attachments tumblr_inline_mqvdlydGCp1qz4rgp • 124 kB • 741 click 2022年3月5日Capture […]
估计阅读时长: 15 分钟https://gcmodeller.org 在这篇博客文章之中,我主要是来详细介绍一下是如何从头开始实现Phenograph单细胞分型算法的。在之前的一篇博客文章《【单细胞组学】PhenoGraph单细胞分型》之中,我们介绍了Phenograph算法的简单原理,以及一个在R语言之中所实现的Phenograph算法的程序包Rphenograph。在这里我主要是详细介绍在GCModeller软件之中所实现的VisualBasic语言版本的Phenograph单细胞分型算法。 Attachments Rphenograph • 236 kB • 814 click 2021年9月20日
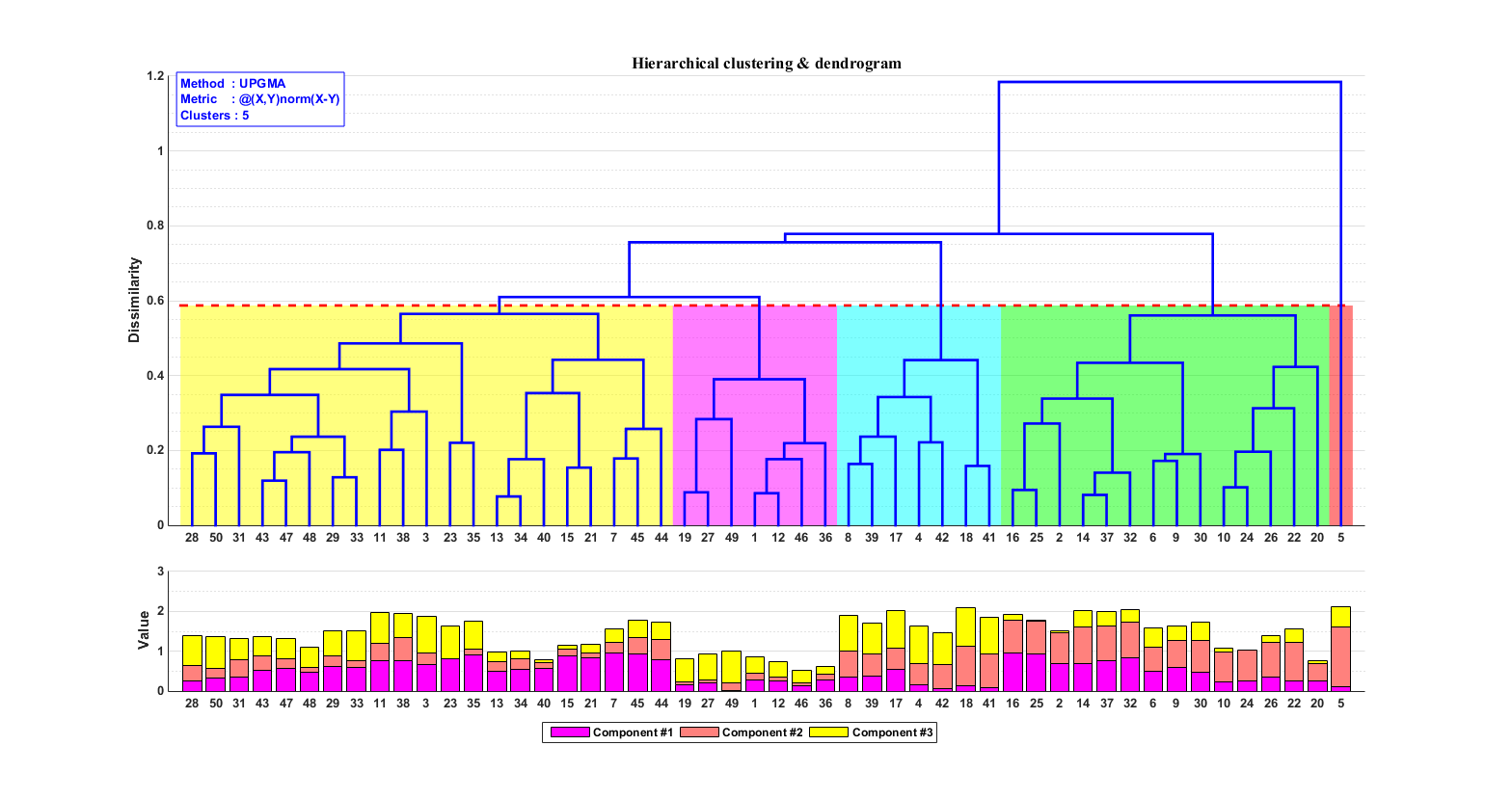
估计阅读时长: 14 分钟https://github.com/xieguigang/sciBASIC 层次聚类通过计算不同类别数据点间的相似度来创建一棵有层次的嵌套聚类树。基于层次聚类分析,我们可以初步可视化我们的一些原始数据: 例如对样本的层次聚类分类,可以让我们了解到样本在分组之间以及分组内的异质性。 对生物序列进行基于相似度的层次聚类分析,我们可以了解到序列之间的相似性程度或者进化关系 Order by Date Name Attachments metabolome • 14 kB • 848 click […]
估计阅读时长: 8 分钟https://github.com/xieguigang/sciBASIC 在进行无监督聚类分析的方法之中,我们在算法代码之中一般会遇到求解与某一个样本数据点最相似的数据点的计算过程。对于这个计算过程,一般而言我们是基于欧几里得距离来完成的。 Order by Date Name Attachments Visual a KDtree Search • 274 kB • 884 […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?