
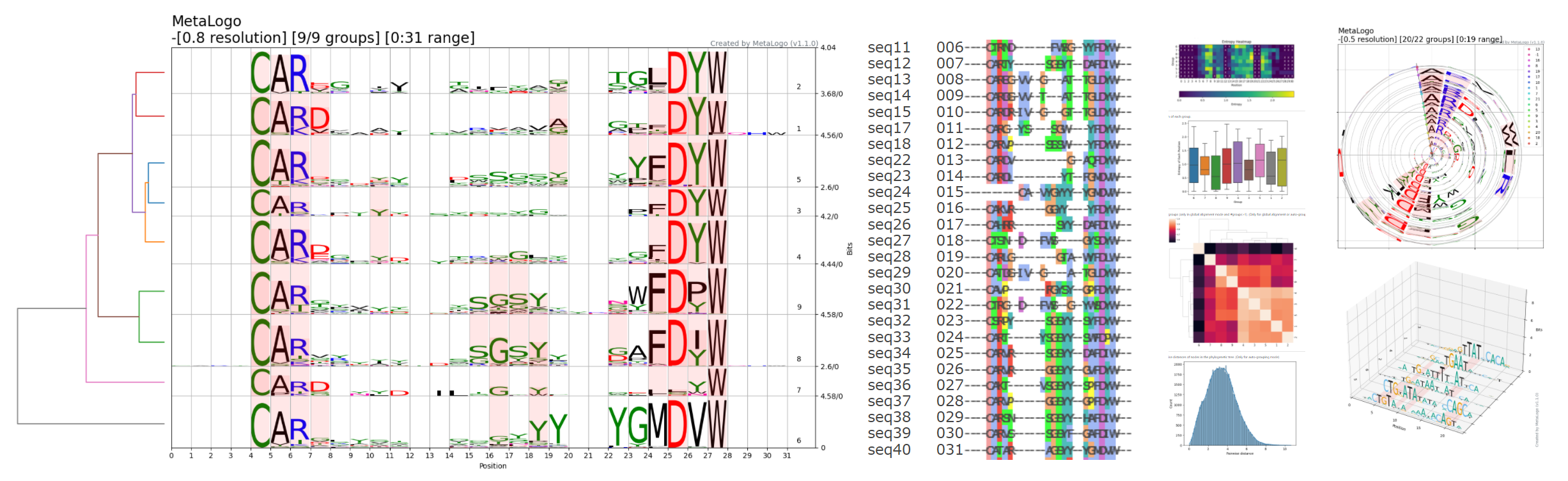
估计阅读时长: 23 分钟Sequence Logo 是一种可视化 DNA 或蛋白质序列保守性的图形表示方法。每个位置(列)上的字母堆叠高度代表该位点的信息含量(以 bits 为单位),而每个字母的高度则与其在该位点出现的频率成正比。高信息量的位置字母堆得高,低信息量的位置则矮甚至接近零。Sequence Logo的绘制遵循信息熵原理,我们可以很直观的通过某一个位置的总高低来了解该处位置的信息含量有多少,高信息量的位置,字母堆的高,一般会出现某一个字符特别高,表明该处非常保守。 位置权重矩阵(Position Weight Matrix, PWM)是描述基因组调控因子结合位点序列模式的核心模型。它通过统计在结合位点序列中每个位置上各核苷酸(或氨基酸)出现的频率,来量化该位置对不同碱基的偏好程度。PWM通常以矩阵形式表示,行对应核苷酸(A、C、G、T/U),列对应序列中的位置,矩阵元素即为该位置该核苷酸相对于背景的权重得分。这一模型简洁且易于计算,因此在转录因子结合位点(TFBS)等调控元件的识别和表征中被广泛采用。 Order by Date Name […]

估计阅读时长: 24 分钟假若现在有两条Fasta序列放在你面前,现在需要你进行这两条Fasta序列的相似度计算分析。如果对于我而言,大学刚毕业刚入门生物信息学的时候,可能只能够想到通过blast比对的方式进行序列相似性计算分析。基于blast比对方式可以找到生物学意义上的序列相似性结果,但是计算的效率会比较低。假设现在让你使用这些序列进行机器学习建模分析,或者基于传统数学意义上的基于相似度的无监督聚类分析的时候,面对这些长度上长短不一的生物序列数据,可能会比较蒙圈,因为传统的数学分析方法都要求我们分析的目标至少应该是等长的向量数据。 Order by Date Name Attachments Fasta-A • 544 kB • 934 click 2023年6月29日visualize • 45 […]

估计阅读时长: 2 分钟Docker镜像信息 GCModeller以R#语言的软件包的形式提供给客户使用,相应的R#语言的分析环境以Docker镜像的形式进行打包盒发布,Docker的基础镜像为ubuntu 22.04。 dotnet环境:.NET 6 R#语言安装位置:/usr/local/bin R#程序包安装列表: 索引 包名称 Github 1 GCModeller https://github.com/SMRUCC/GCModeller 2 REnv https://github.com/rsharp-lang/R-sharp […]
估计阅读时长: 7 分钟一般而言,进行全基因组的转录表达调控网络的建立,我们需要基于两个数据结果来完成: 目标基因的转录调控位点信息(Motif搜索结果,构成网络之中的节点) 转录调控位点相应的转录调控因子(Motif位点相关的转录调控因子,构成网络之中的边连接) Order by Date Name Attachments Xor • 271 kB • 1017 click 2022年6月11日An […]
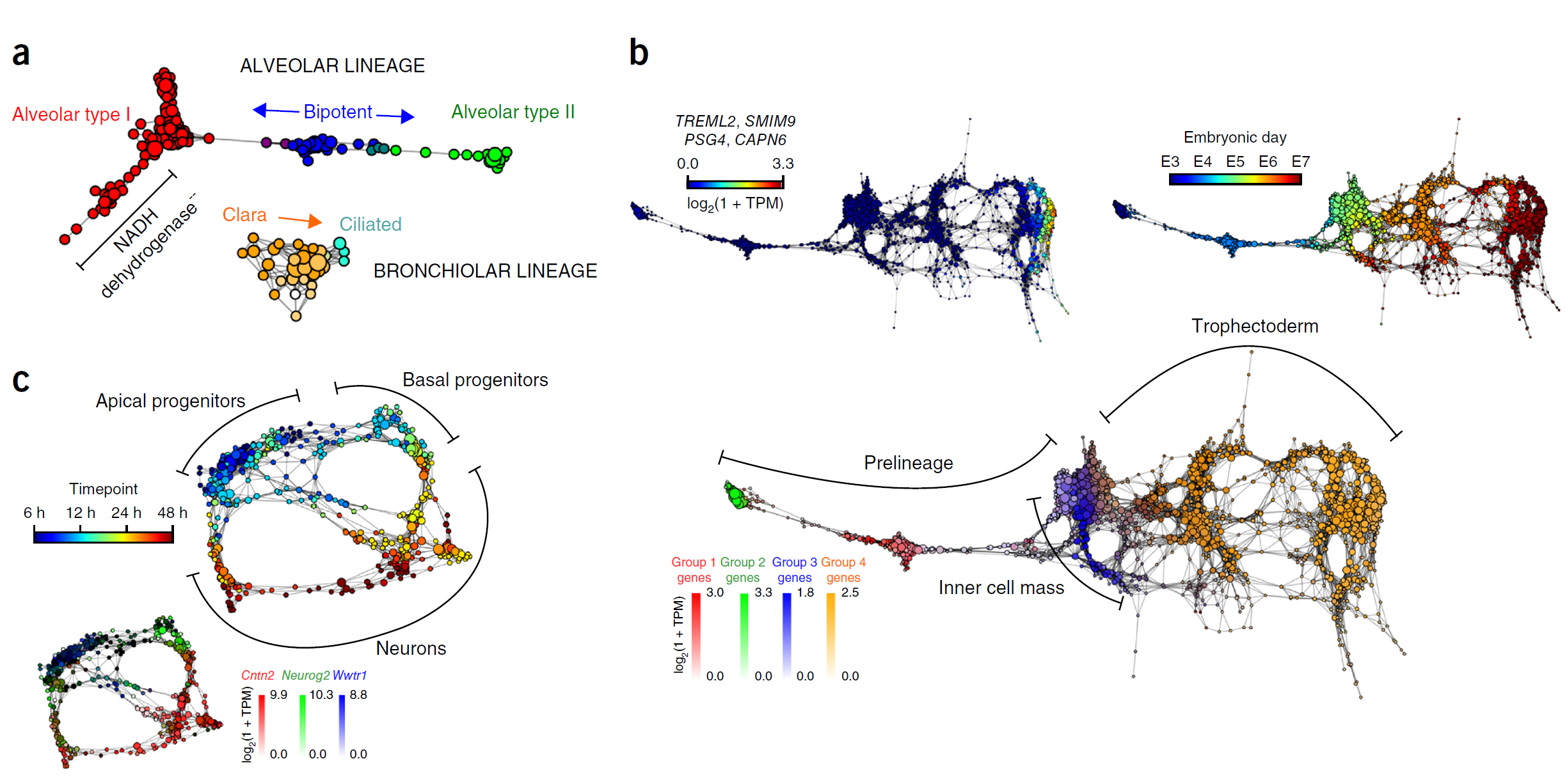
估计阅读时长: 14 分钟单细胞分析方法学习文献打卡记录: 【单细胞组学】PhenoGraph单细胞分型 【单细胞分析方法】VeTra:基于RNA速度的轨迹推断工具 【单细胞分析方法】单细胞图嵌入 Order by Date Name Attachments Cellular populations during motor neuron differentiation • […]
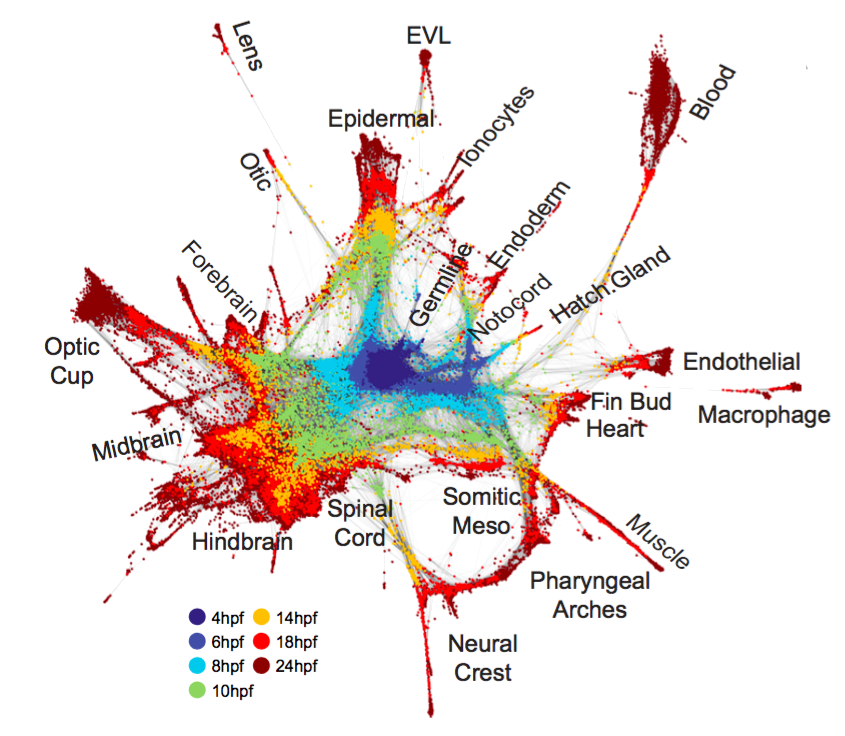
估计阅读时长: 7 分钟Assembles a manifold that is defined through a series of overlapping, locally-defined PCA subspaces. Non-mutual k-nearest-neighborhoods […]
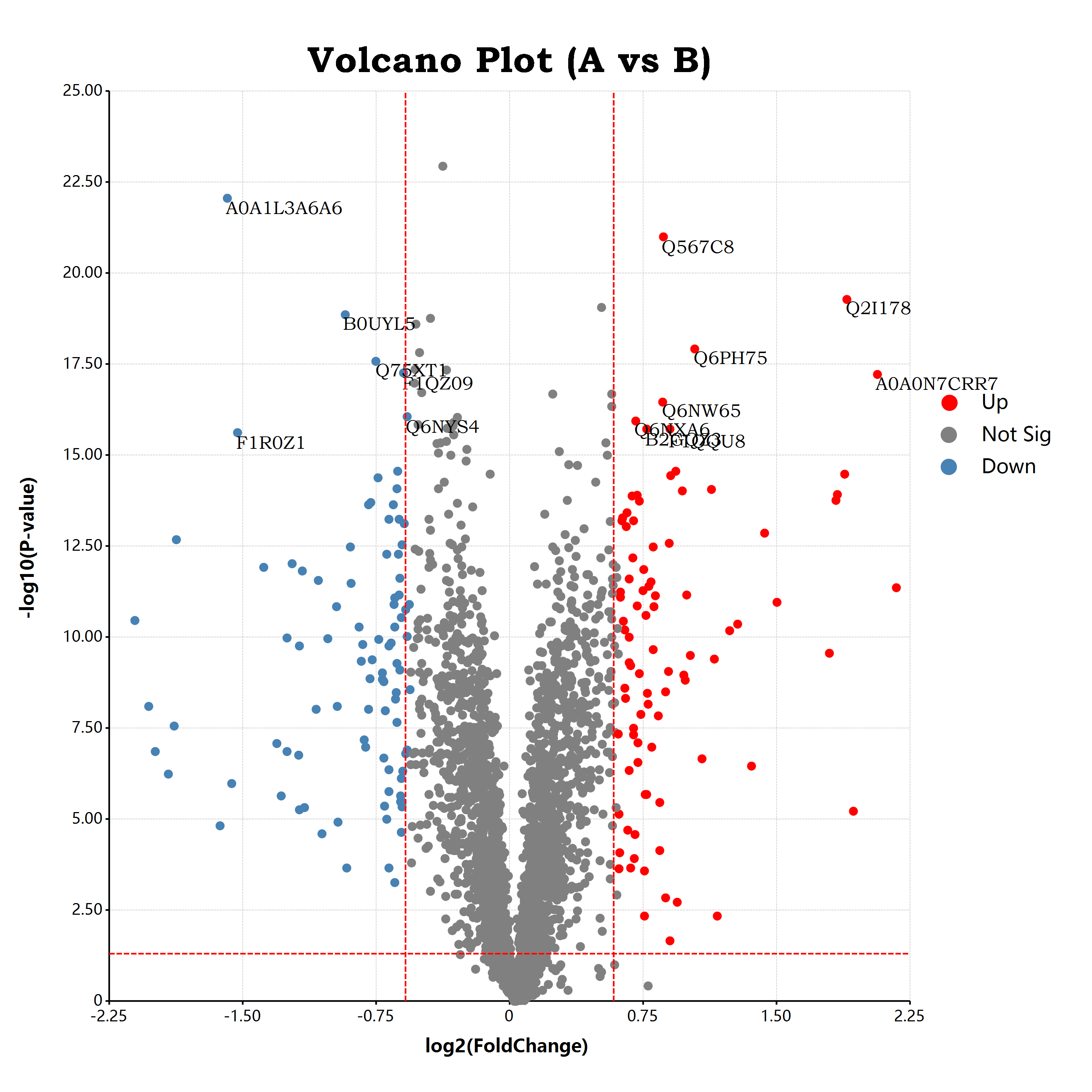
估计阅读时长: 17 分钟https://github.com/rsharp-lang/ggplot 接上一篇博客文章中谈到,我们已经通过R#语言之中的ggplot程序包绘制出了一个可以使用的火山图。在这里,我们将会通过在火山图上添加更多的可视化元素来为大家介绍R#语言之中的ggplot程序包的进阶使用方式。 Order by Date Name Attachments volcano • 651 kB • 1105 click 2021年10月9日volcano • […]
估计阅读时长: 11 分钟https://github.com/rsharp-lang/ggplot 在生物信息学中的组学数据分析领域内,有一个非常常见的数据可视化图表:应用于可视化两两组别比对结果的火山图。在火山图之中,X坐标轴一般是log2FC,纵坐标Y轴,则一般是t检验的pvalue的-log10转换之后的值。由于fold change有大于1的值,A/B大于1,表示A的表达量高于B的表达量,反之小于一表示A的表达量低于B的表达量。这样子fold change经过log2转换之后,就会出现负数,散点一般呈轴对称分布在X=0的位置周围。这样子绘制出来的散点图就有点类似于火山喷发的样子了。 Order by Date Name Attachments a679af1eb9ffbfbad48c18d563ea51f3 • 45 kB • 1009 click […]
估计阅读时长: 15 分钟https://gcmodeller.org 在这篇博客文章之中,我主要是来详细介绍一下是如何从头开始实现Phenograph单细胞分型算法的。在之前的一篇博客文章《【单细胞组学】PhenoGraph单细胞分型》之中,我们介绍了Phenograph算法的简单原理,以及一个在R语言之中所实现的Phenograph算法的程序包Rphenograph。在这里我主要是详细介绍在GCModeller软件之中所实现的VisualBasic语言版本的Phenograph单细胞分型算法。 Attachments Rphenograph • 236 kB • 1058 click 2021年9月20日
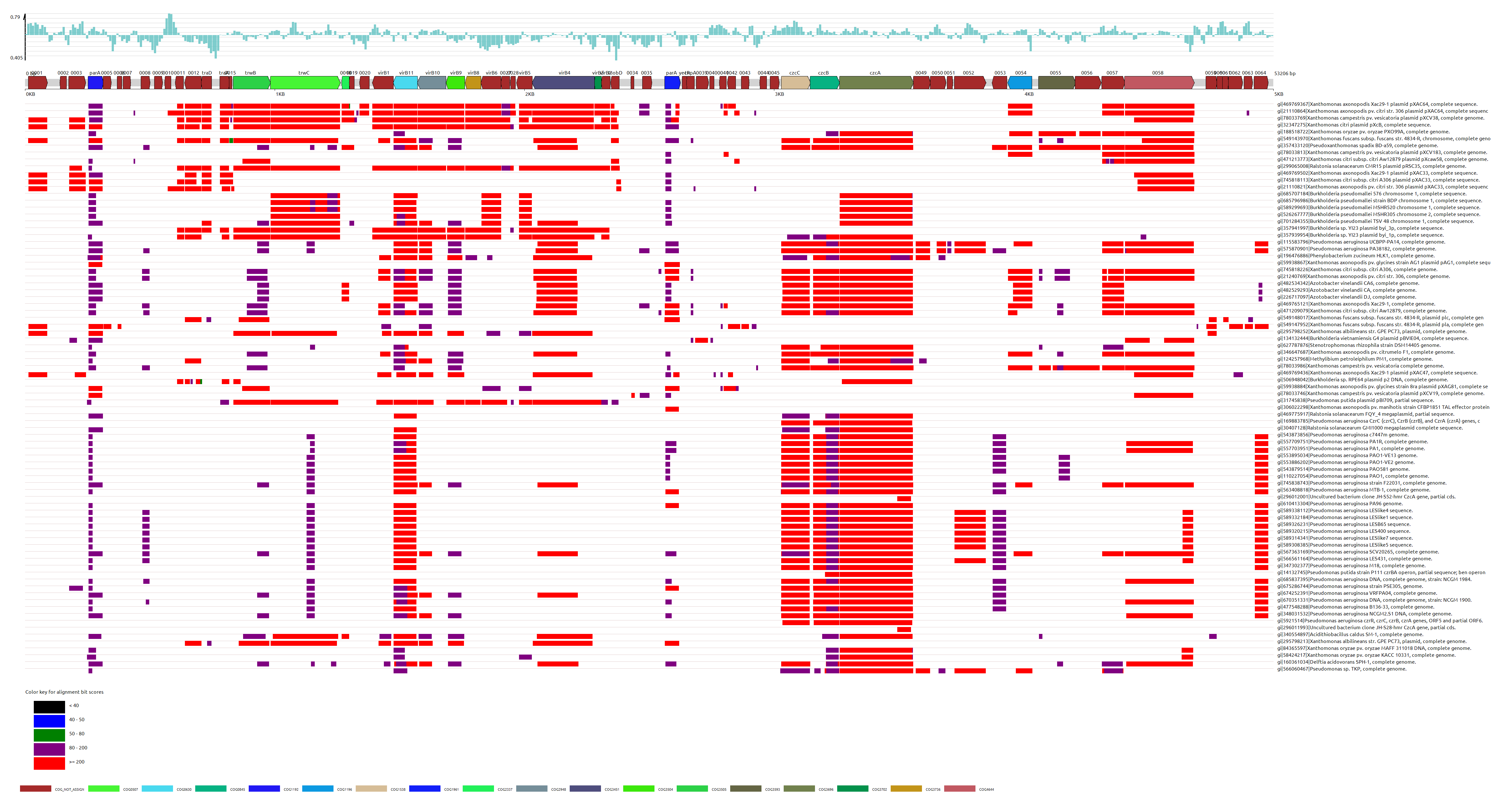
估计阅读时长: 14 分钟在基因组学研究中,将新测序的基因或者针对目标基因组进行基于KEGG代谢通路体系的虚拟细胞建模,都会需要将目标基因组与已知功能基因进行比对注释。KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库通过其KEGG Orthology (KO)系统,为基因功能注释提供了一个强大的平台。KO系统将功能上保守的直系同源基因归为一类,每个KO条目(K编号)代表一个直系同源基因群,这些基因在不同物种中通常执行相似的生物学功能。因此,将新基因的序列与KEGG数据库中的已知基因进行比对,可以推断其可能的KO编号,从而将其功能映射到KEGG通路图或功能层级中。 Order by Date Name Attachments kegg_overview • 313 […]

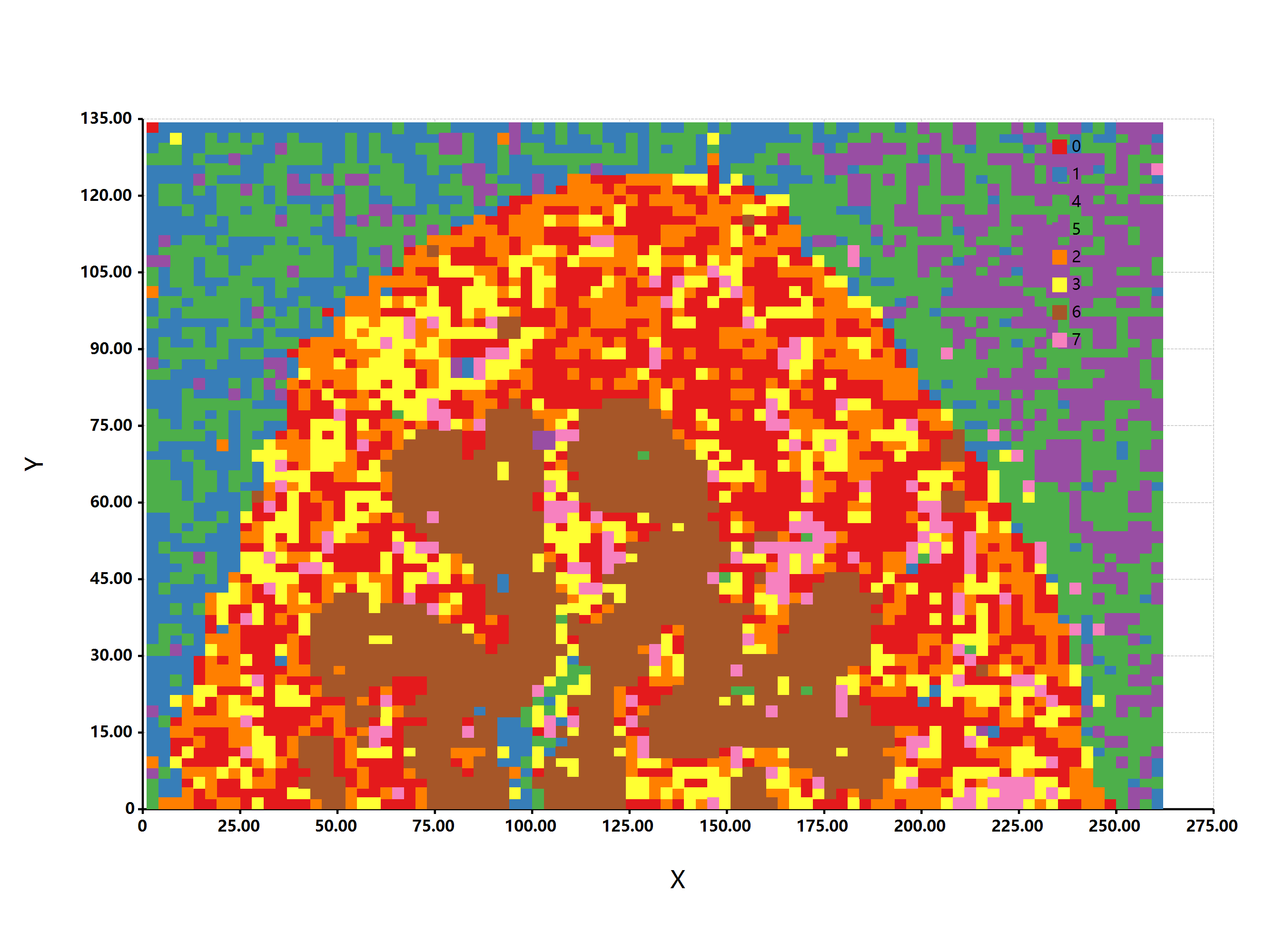
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?