
估计阅读时长: 6 分钟大家好呀,今天的这篇文章主要是为了回答在B站上的一位小伙伴的请求 Order by Date Name Attachments render-parameters • 18 kB • 207 click 2023年10月15日view-umap • 427 […]

估计阅读时长: 7 分钟假若现在有两条Fasta序列放在你面前,现在需要你进行这两条Fasta序列的相似度计算分析。如果对于我而言,大学刚毕业刚入门生物信息学的时候,可能只能够想到通过blast比对的方式进行序列相似性计算分析。基于blast比对方式可以找到生物学意义上的序列相似性结果,但是计算的效率会比较低。假设现在让你使用这些序列进行机器学习建模分析,或者基于传统数学意义上的基于相似度的无监督聚类分析的时候,面对这些长度上长短不一的生物序列数据,可能会比较蒙圈,因为传统的数学分析方法都要求我们分析的目标至少应该是等长的向量数据。 Order by Date Name Attachments Fasta-A • 544 kB • 185 click 2023年6月29日visualize • 45 […]

估计阅读时长: 2 分钟Docker镜像信息 GCModeller以R#语言的软件包的形式提供给客户使用,相应的R#语言的分析环境以Docker镜像的形式进行打包盒发布,Docker的基础镜像为ubuntu 22.04。 dotnet环境:.NET 6 R#语言安装位置:/usr/local/bin R#程序包安装列表: 索引 包名称 Github 1 GCModeller https://github.com/SMRUCC/GCModeller 2 REnv https://github.com/rsharp-lang/R-sharp […]
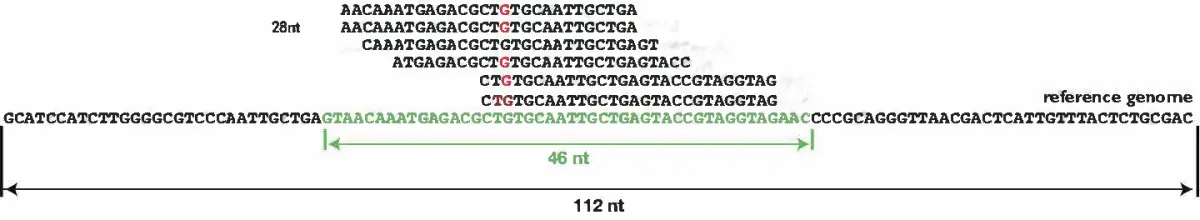
估计阅读时长: 11 分钟给定一组n个字符串数组,找到包含给定集合中每个字符串的最小字符串作为子字符串。我们可以假设这个字符串数组中没有字符串是另一个字符串的子字符串。那么基于上面的描述,我们就可以得到下面所示的问题求解目标: let arr[] = ["catg", "ctaagt", "gcta", "ttca", "atgcatc"] // output: gctaagttcatgcatc 上面的问题描述实际上是一个最短超字符串问题(shortest common superstring) Order […]

估计阅读时长: 6 分钟CentOS查看系统版本信息 cat /etc/redhat-release # CentOS Stream release 8 cat /proc/version # Linux version 4.18.0-489.el8.x86_64 (mockbuild@x86-05.stream.rdu2.redhat.com) (gcc […]

估计阅读时长: 5 分钟https://github.com/xieguigang/scale_colour_genshin 在用R绘图时,颜色设置是美化过程中不可缺少的一步。在实际绘图时,一般不会一一手动寻找合适的颜色,而是通过一些R包、网站提供好的,美观的颜色组合,即调色板(palette),可供使用。在这里介绍一种通过提取图片主题色的方法来为我们自动生成画图所用的颜色板数据。 Order by Date Name Attachments 383807b4 • 132 kB • 217 click 2023年4月8日faruzan • […]

估计阅读时长: 9 分钟因为一种单一的编程语言并不会覆盖到所有的适用场景的原因,在一个软件工程项目之中,采用多种语言进行混合编程是一种很常见的协作方式。例如,脚本化的语言,其非常适合于进行最顶层的应用开发,就像胶水一样用于将各种组件进行粘贴,但是脚本化的语言自身因为是基于其他的语言所构建,所以执行效率一般不会太好。对于底层组件,我们一般就会需要使用静态编译类型的非托管语言创建用于高性能数据处理的模块。对于这种需求的底层模块,我们一般可以采用C/C++/Rust来编写。 Order by Date Name Attachments rust • 162 kB • 208 click 2023年3月25日dyn-load • 67 […]
估计阅读时长: 8 分钟https://github.com/rsharp-lang/NRRD NRRD(Nearly Raw Raster Data)是一种用于存储类似于热图成像数据的文件格式。其实我们可以将NRRD看作为类似于bitmap之类的未压缩的原始光栅图像文件。只要我们有对应的解码方式,我们就可以像查看普通图片文件一样查看NRRD文件。 Order by Date Name Attachments raster__238 • 61 kB • 220 […]
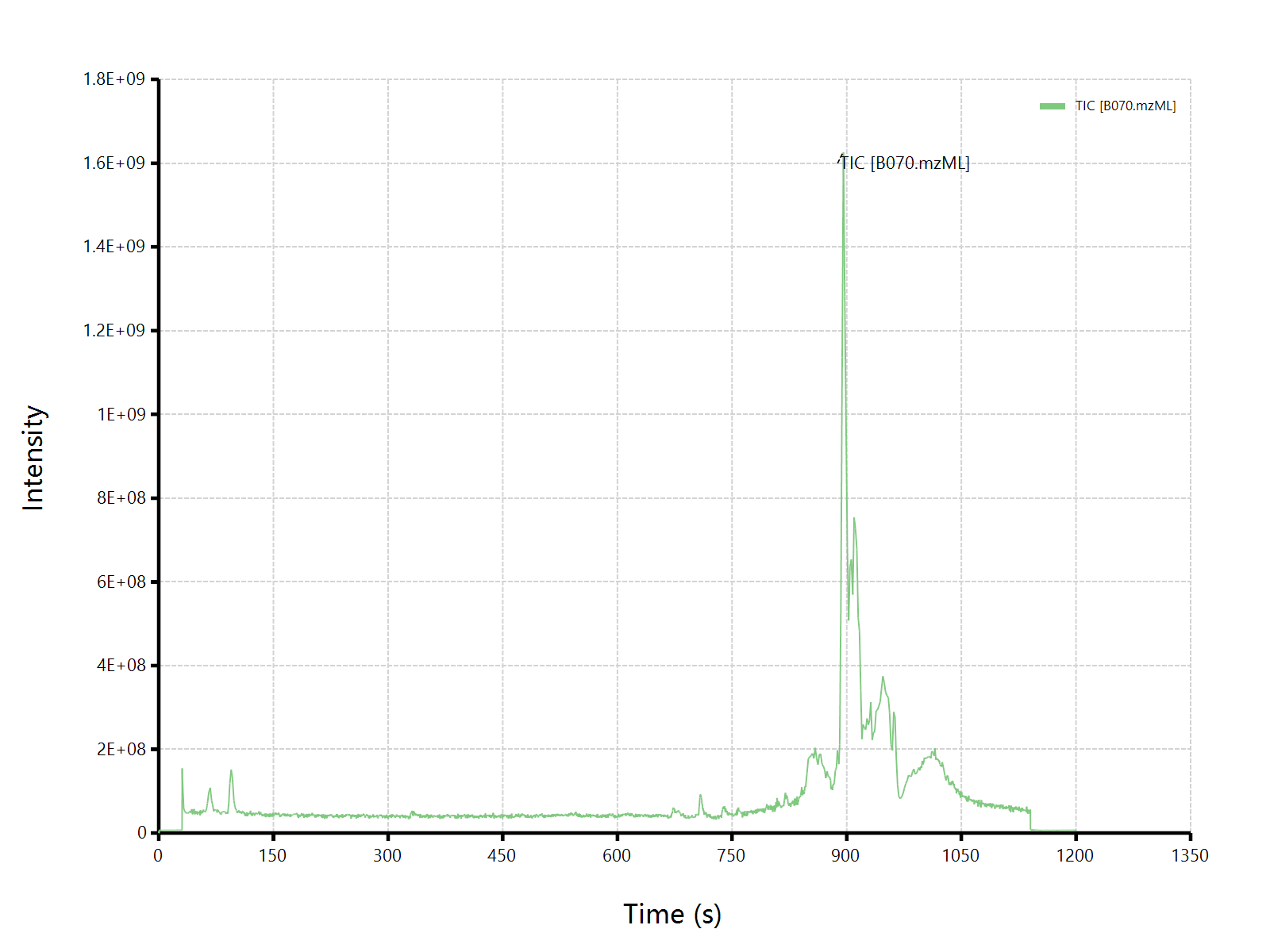
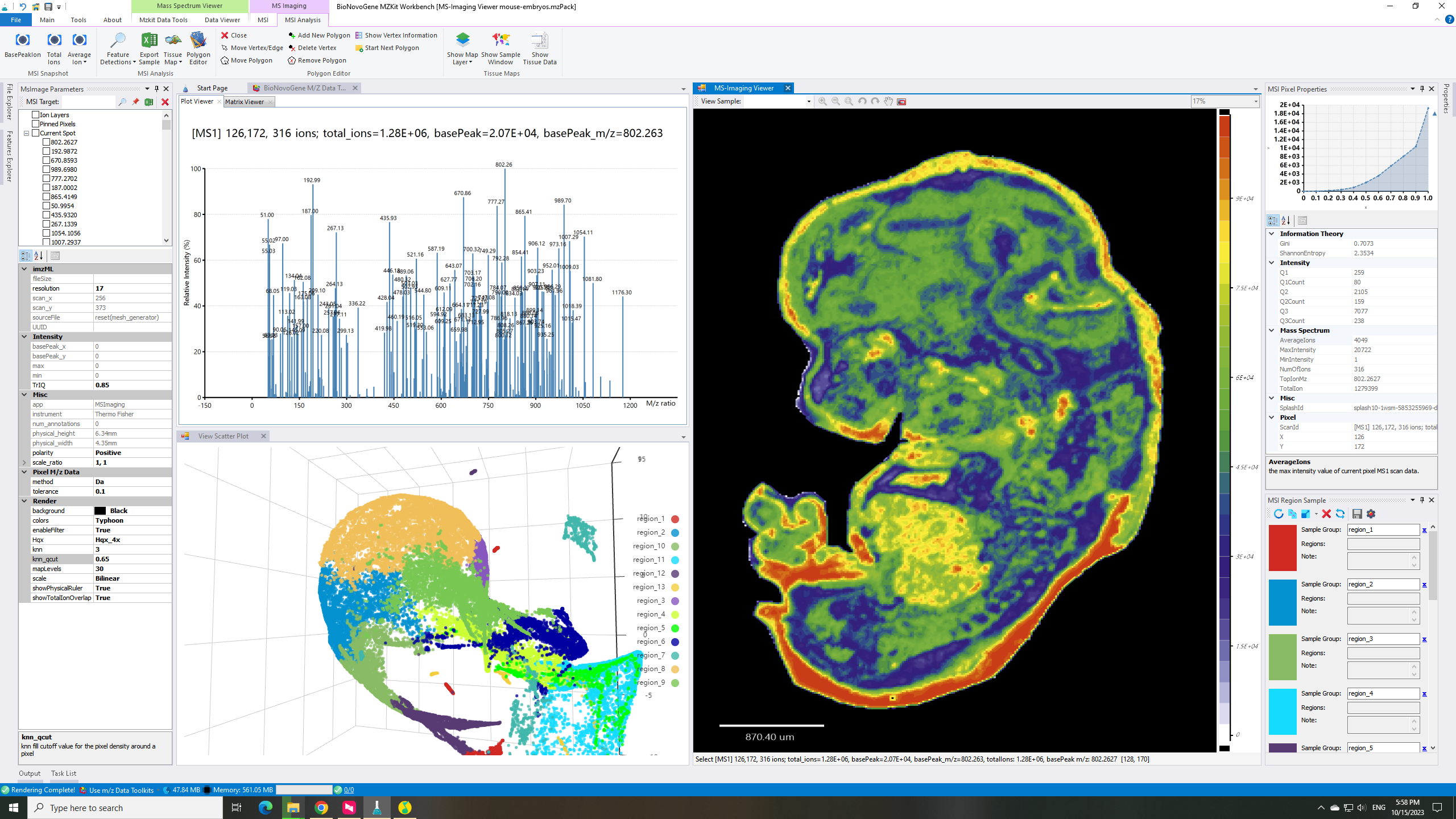
估计阅读时长: 5 分钟在BILIBILI上观看视频:《【BioNovoGene Mzkit教程】代谢组学原始数据处理基础》 最近我在B站的视频页面下发现了这样的一条评论,面对质谱数据分析领域内的初学者的求教,其实自己也是非常的诚惶诚恐的。因为在视频中所使用的脚本语言是自己开发的一门新语言,所以可能给一些初学者造成了一部分的困扰哈哈😅😄😅😅。首先先对这个粉丝说一声抱歉哈。 针对上述的提问,我的回答大概是有以下的几点: Order by Date Name Attachments question_20230223 • 17 kB • 177 click […]
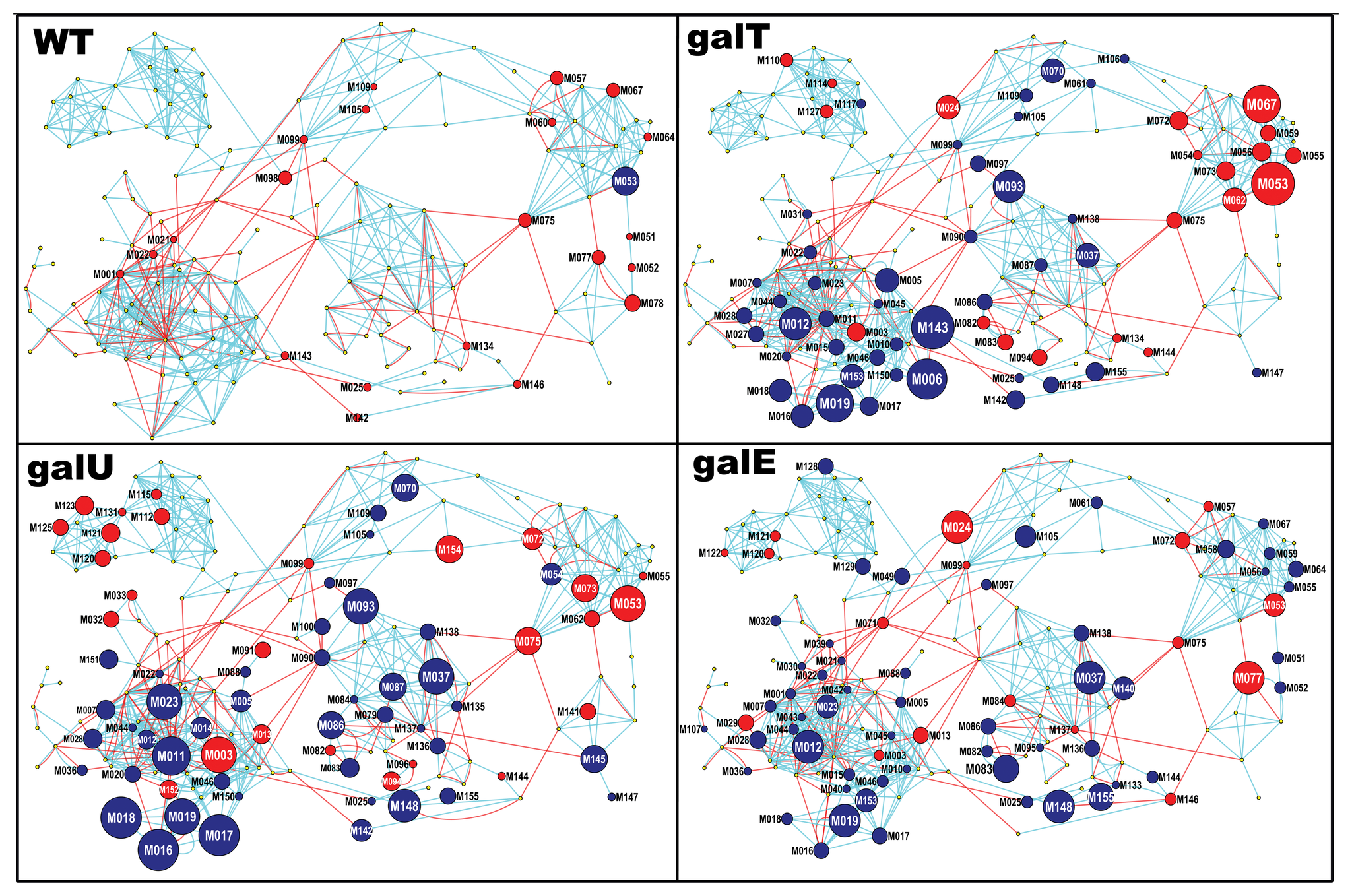
估计阅读时长: 5 分钟在工作之中可能会遇到需要进行两个网络图对象之间的相似度计算的情形:例如在质谱数据分析的化学信息学计算工作之中,我们在解析SMILES字符串得到分子图之后,可以基于图相似度比较计算方法来比较计算两个代谢物分子图之间的结构上的相似度。 Attachments pone.0078360.g003 • 2 MB • 316 click 2022年8月6日https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0078360

Hello There. I found your website through the usage of msn. This is a very smartly written article. I will…
[…] […]
[…] […]
[…] 《为大语言模型运行添加工具调用》 […]
[…] 《从头创建一个DeepSeek客户端》 […]