
文章阅读目录大纲
 https://gcmodeller.org/
https://gcmodeller.org/
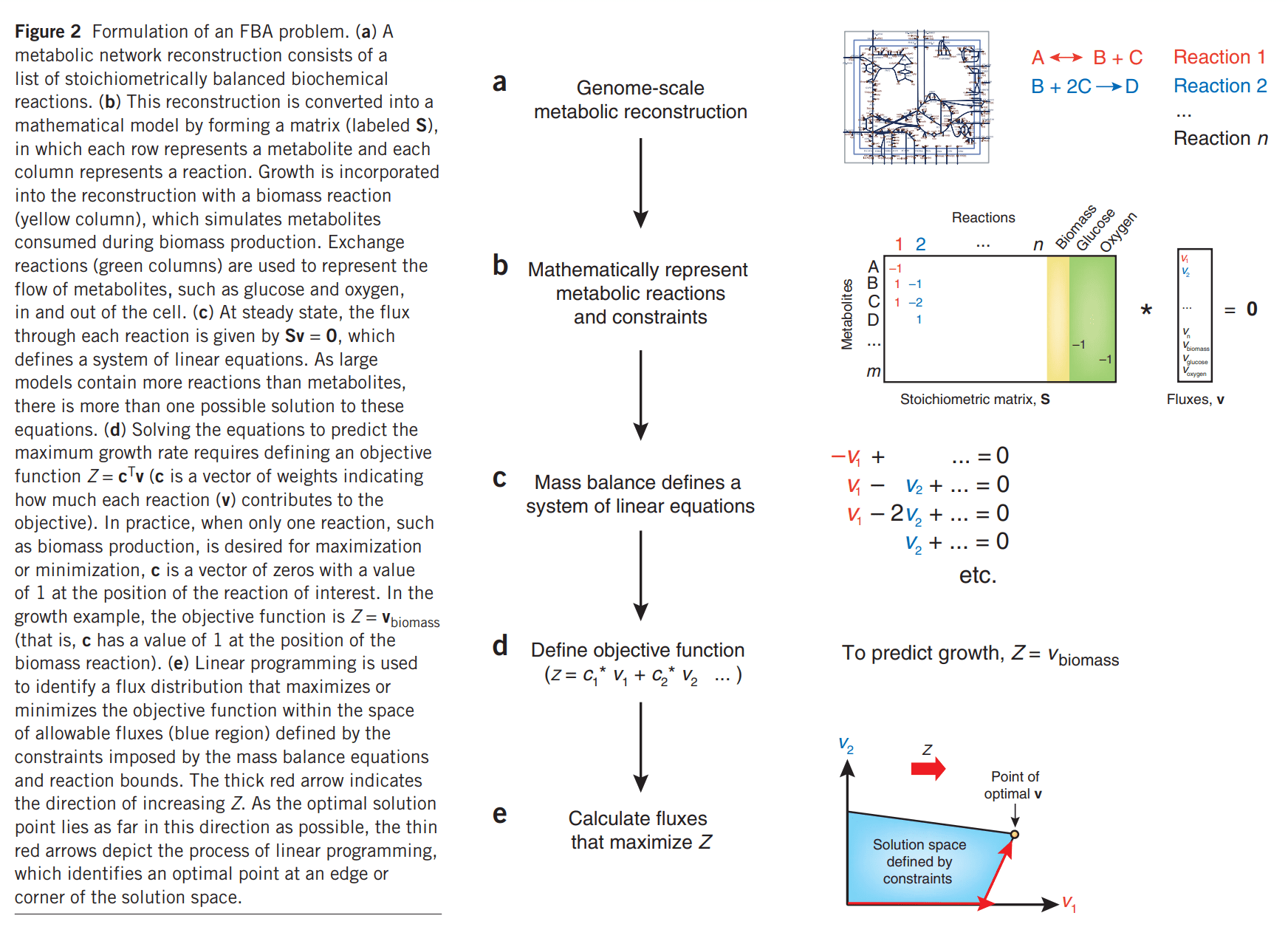
流平衡分析(flux balance analysis)是一种可以用来构建和模拟分析基因组级别的代谢网络的数学方法。流平衡分析是系统生物学(system biology)的一个重要的分析手段。不同于以湿实验的代谢通量分析(metabolic flux analysis, MFA),FBA是用数学方法对代谢网络里的代谢流进行拟合分析。
前言
在这里顺着公司部门组会内的文献分享的作业,尝试通过自己所编写的GCModeller软件进行文献中的FBA分析过程的复现:
Predicting the metabolic capabilities of Synechococcus elongatus PCC 7942 adapted to different light regimes. Metab Eng. 2019 March; 52: 42–56. doi:10.1016/j.ymben.2018.11.001.
自己所进行分享的文献是2019年发表在代谢工程(Metab Eng)期刊上的一篇关于蓝藻的代谢研究。首先来稍微了解一下代谢工程这个期刊的一些基本信息:
Metabolic Engineering 期刊介绍
2021年影响因子/JCR分区:9.783/Q2
大类学科及分区:
工程技术 1区
是否TOP期刊:是
小类学科及分区:
BIOTECHNOLOGY & APPLIED MICROBIOLOGY
生物工程与应用微生物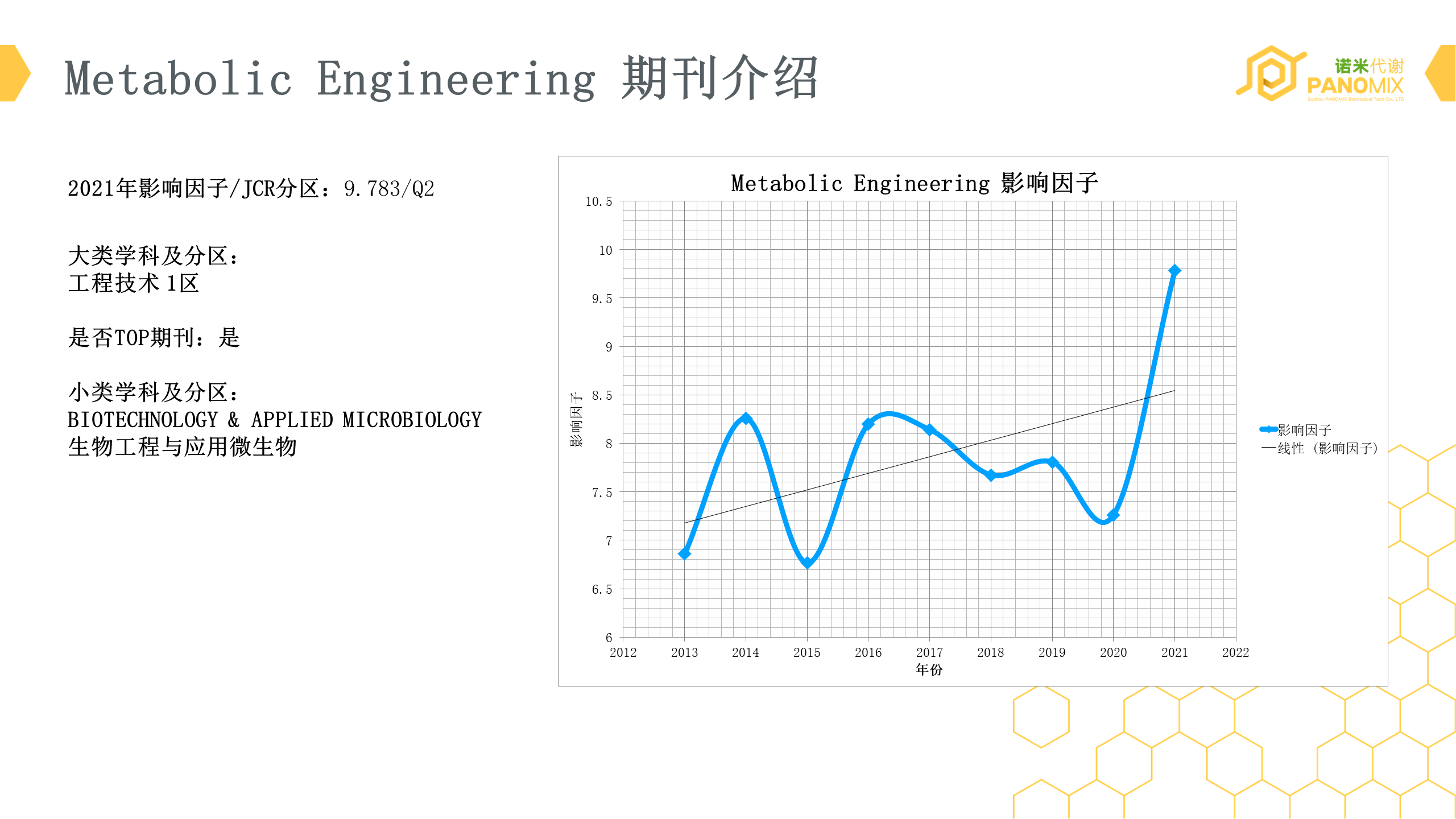
文章简单解读
基于GCModeller的代谢网络建模
在进行FBA代谢网络分析之前,我们需要对目标基因组重建代谢网络。这样子我们才可以依据所重建出来的代谢网络构建出线性规划计算所需要的相关系数矩阵。一般而言,重建代谢网络我们主要是进行全基因组注释,主要的注释对象是酶的编号。这样子当我们的目标基因组蛋白具有了酶编号之后,就可以依据酶编号从各种代谢数据库中查询获取得到相应的酶促代谢反应过程列表。依据所得到的代谢反应列表完成代谢网络的可视化以及线性规划矩阵的建立。
因为就目前而言,日本的KEGG数据库是比较权威经典的代谢功能相关的数据库,在其中记录了各种酶,酶促代谢反应,相关的参考序列信息,各种信息与数据很齐全。所以目前最常见的全基因组注释方法,我们一般是通过KEGG数据库注释来完成的。关于通过KEGG数据库进行代谢网络的重建,大家可以观看GCModeller之前的一个课程视频:《【GCModeller教程】KEGG代谢途径注释原理》。
基础准备工作
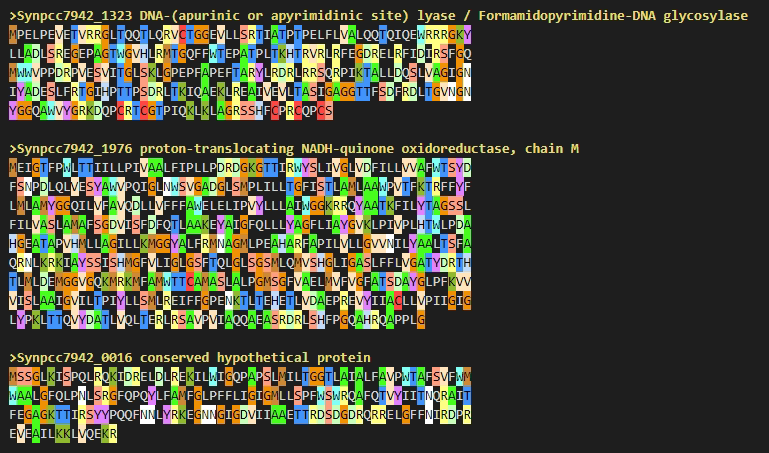
在这里我们以非模式生物分析的流程来进行蓝藻的代谢网络分析。首先我们会需要拥有非模式生物分析流程最原始的数据:蛋白序列。在这里我通过genbank数据库文件来导出蛋白序列。
imports ["GenBank", "bioseq.fasta"] from "GCModeller::seqtoolkit";
imports "blast+" from "seqtoolkit";
`${@dir}/../ncbi-genomes-2021-09-06/GCA_000012525.1_ASM1252v1_genomic.gbff`
|> read.genbank
|> (function(gbff) {
const prot_fsa as string = `${@dir}/../annotations/proteins.fasta`;
print("previews of the genome information:");
print(gbff);
protein.fasta(gbff)
|> write.fasta(
file = prot_fsa,
lineBreak = 60
)
;
makeblastdb(prot_fsa, dbtype = "prot");
})
;执行上面的一段R#脚本,可以得到一个蛋白序列文件:
接着,会需要从整个UniProt数据库之中导出具有KEGG直系同源编号的所有蛋白序列。(在旧版本的UniProt数据库之中,蛋白序列是会存在着人工注释的KO编号的,但是最近(2021.07)我更新过UniProt数据库后发现原来存在的KO编号都消失了,不太清楚是不是因为版权的原因导致UniProt数据库删除了所有KEGG直系同源的编号记录,还好我这里还存有旧版本的UniProt数据库备份。)
imports ["uniprot", "bioseq.fasta"] from "GCModeller::seqtoolkit";
imports "blast+" from "seqtoolkit";
setwd(@dir);
write.fasta(file = "./UniProt_KO.fasta") {
# exact KEGG orthologous reference
# sequence database
# for run KO mapping
[
`./uniprot_sprot.xml`,
`./uniprot_trembl.xml`
]
|> open.uniprot
|> protein.seqs(KOseq = TRUE)
;
}
makeblastdb("./UniProt_KO.fasta", dbtype = "prot");基因组功能注释
我们在这里主要是通过COG注释以及KO注释来进行蓝藻基因组的注释分析。在这里我们通过双向最佳比对的方法来进行蛋白功能注释分析,以下展示的是基于NCBI blast+软件包的流程封装脚本:
imports ["blast+", "annotation.workflow"] from "GCModeller::seqtoolkit";
const query = `${@dir}/proteins.fasta`;
const uniprot = `${@dir}/UniProt_KO.fasta`;
#' run ncbi localblast+
#'
#' @param query the filepath of the query fasta
#' @param ref the filepath of the reference fasta
#' @param fileName the file basename of the result table file
#'
#' @return the file path of the resulted SBH table file.
#'
const runBlast as function(query, ref, fileName) {
const filepath = `${@dir}/paralogs/${fileName}.csv`;
blastp(
query = query,
db = ref,
evalue = 1E-5,
n_threads = 60
)
|> read.blast(type = "prot", fastMode = TRUE)
|> blasthit.sbh(idetities = 0.5, coverage = 0.8, topBest = TRUE, keepsRawName = FALSE)
|> stream.flush(stream = open.stream(
file = filepath,
type = "SBH"
))
;
filepath;
}
const forward = open.stream(runBlast(query, uniprot, "forward"), ioRead = TRUE);
const reverse = open.stream(runBlast(uniprot, query, "reverse"), ioRead = TRUE);
# create KO mappng result table
# via BBH orthologous hybrids SBH paralogs
# with kegg BHR score assignment algorithm
blasthit.bbh(forward, reverse, algorithm = "Hybrid-BHR")
|> stream.flush(stream = open.stream(
file = `${@dir}/UniProt_KO.csv`,
type = "BBH"
))
;在这里我们使用的是KEGG的KAAS自动化注释系统中的BHR评分算法来进行直系同源的注释分析。关于BHR评分算法进行直系同源注释的更加详细的讲解,大家可以阅读我的这篇博客文章:《KEGG的BHR评分注释直系同源》,在这里不做过多的赘述。
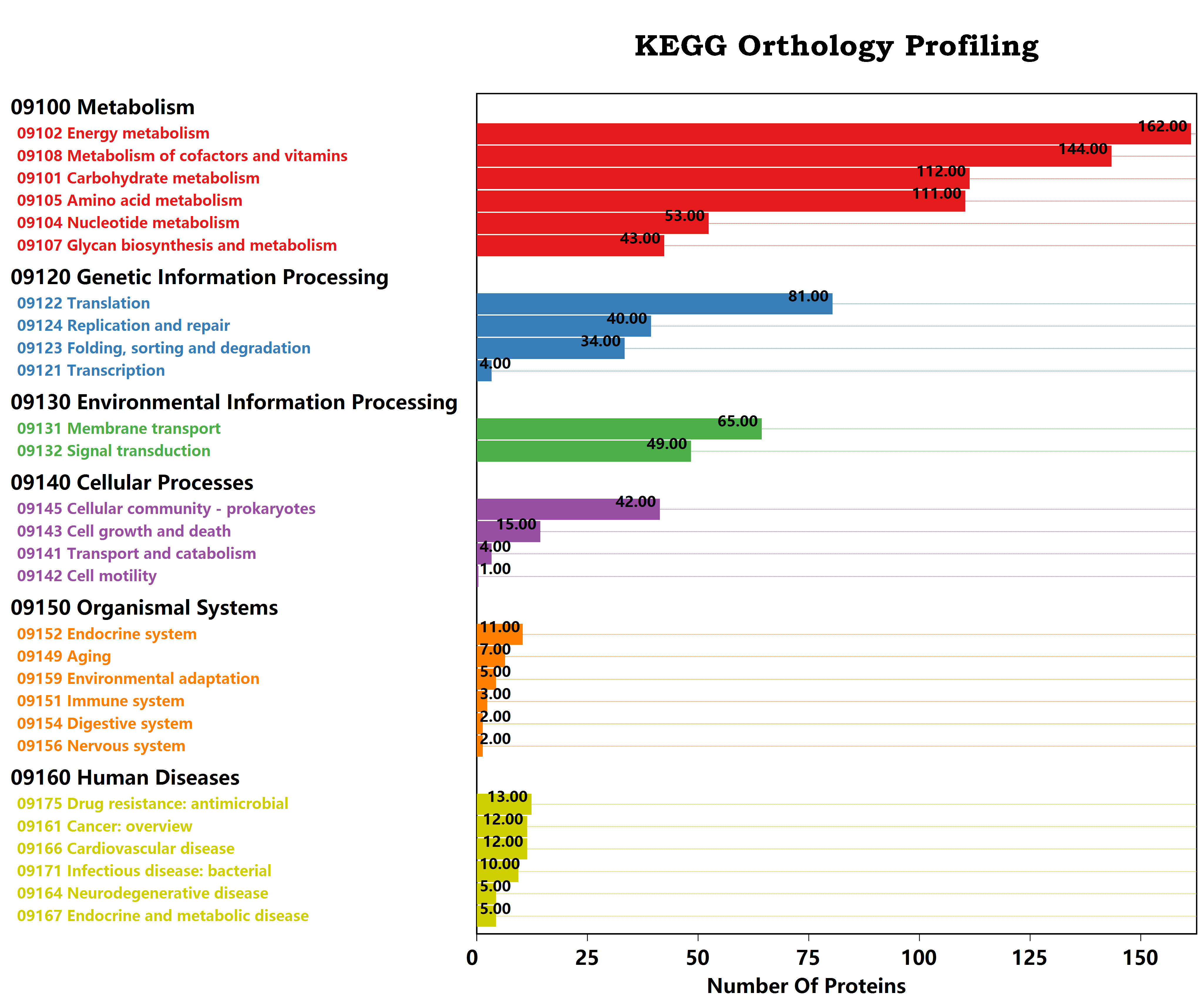
KEGG直系同源注释结果可视化
在这里我们使用条形图对KEGG注释统计结果进行可视化:
require(GCModeller);
imports "profiles" from "kegg_kit";
imports "visualPlot" from "visualkit";
setwd(@dir);
bitmap(file = "../annotations/KO/profiles.png") {
read.csv("../annotations/KO/bbh_annotation.csv")[, "term"]
|> unique
|> which(id -> id != "")
|> KO.map.profiles
|> kegg.category_profile(top = 6, sort = TRUE)
|> plot(
title = "KEGG Orthology Profiling",
axis.title = "Number Of Proteins",
size = [4100,3400]
);
}重建KEGG代谢网络
那,我们在进行完KO注释工作之后,实际上就相当于对蓝藻基因组之中的每一个基因都分配有了一个KEGG数据库之中的直系同源编号了。因为在KEGG数据库之中,KO编号实际上很大程度上是与酶一一对应的。所以我们就可以依据所注释得到的KO编号找到KO编号所对应的酶促生化反应。基于整个基因组的KO编号列表,就可以从整个KEGG数据库中的代谢反应列表中得到一个酶促生化反应的子集。基于这个代谢反应列表中的代谢物作为网络节点,代谢反应构成网络的边链接,我们就可以构建出一个基于KEGG数据库重建的蓝藻代谢网络。
下面是一个GCModeller软件之中的KEGG代谢反应过程的模型数据示例,用来说明如何将我们所注释到的KEGG直系同源编号关联上对应的代谢反应:
<?xml version="1.0" encoding="utf-16"?>
<reaction xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance" xmlns:xsd="http://www.w3.org/2001/XMLSchema" xmlns:KO="http://GCModeller.org/core/KEGG/Model/OrthologyTerm.xsd" xmlns="http://GCModeller.org/core/KEGG/DBGET/Reaction.xsd">
<!--
model: SMRUCC.genomics.Assembly.KEGG.DBGET.bGetObject.Reaction
assembly: SMRUCC.genomics.Core, Version=1.0.0.0, Culture=neutral, PublicKeyToken=null
version: 1.0.0.0
built: 1/1/2000 12:00:00 AM
md5: 45badb514ba4a9978deadd60d1690c97
timestamp: 8/18/2021 5:26:04 PM
-->
<ID>R00623</ID>
<commonNames>primary_alcohol:NAD+ oxidoreductase</commonNames>
<def>Primary alcohol + NAD+ <=> Aldehyde + NADH + H+</def>
<equation>C00226 + C00003<=>C00071 + C00004 + C00080</equation>
<enzymes>
<string>1.1.1.1</string>
<string>1.1.1.71</string>
</enzymes>
<comment>general reaction
NADP+ (ec 1.1.1.71, see R00625)</comment>
<pathway>
<data name="rn00071">Fatty acid degradation</data>
</pathway>
<module />
<orthology size="9">
<KO:terms name="K00001" value="alcohol dehydrogenase [EC:1.1.1.1]">/entry/K00001</KO:terms>
<KO:terms name="K00121" value="S-(hydroxymethyl)glutathione dehydrogenase / alcohol dehydrogenase [EC:1.1.1.2841.1.1.1]">/entry/K00121</KO:terms>
<KO:terms name="K04072" value="acetaldehyde dehydrogenase / alcohol dehydrogenase [EC:1.2.1.101.1.1.1]">/entry/K04072</KO:terms>
<KO:terms name="K13951" value="alcohol dehydrogenase 1/7 [EC:1.1.1.1]">/entry/K13951</KO:terms>
<KO:terms name="K13952" value="alcohol dehydrogenase 6 [EC:1.1.1.1]">/entry/K13952</KO:terms>
<KO:terms name="K13953" value="alcohol dehydrogenase, propanol-preferring [EC:1.1.1.1]">/entry/K13953</KO:terms>
<KO:terms name="K13954" value="alcohol dehydrogenase [EC:1.1.1.1]">/entry/K13954</KO:terms>
<KO:terms name="K13980" value="alcohol dehydrogenase 4 [EC:1.1.1.1]">/entry/K13980</KO:terms>
<KO:terms name="K18857" value="alcohol dehydrogenase class-P [EC:1.1.1.1]">/entry/K18857</KO:terms>
</orthology>
<class>
<data name="RC00001">C00003_C00004</data>
<data name="RC00050">C00071_C00226</data>
</class>
<DBLink DBName="RHEA" Entry="10739">https://www.rhea-db.org/reaction?id=10739</DBLink>
</reaction>可以看得到,在每一个KEGG数据库之中的代谢反应模型文件之中,都会存在有一个KO编号列表用来关联这个代谢反应过程到对应的代谢酶蛋白上。接着呢,我们可以对equation字段做出解析,得到代谢物列表以及相对应的化学计量数。通过这些信息,我们就可以依照KO编号的注释结果完成整个代谢网络的重建工作了。得到了代谢网络之后,就可以依照对equation字段信息的解析得到FBA计算分析所需要的系数矩阵。
下面的一段代码就是通过我们前面通过KEGG直系同源编号注释得到的id列表进行代谢网络的重建:
FBA模型计算分析
FBA方法也算是一种很经典的代谢优化分析方法了。
FBA分析简单介绍
FBA分析的一些相关知识库信息的建立,大家可以阅读之前写的两篇博客文章:《【数据可视化】基于必应学术搜索的知识网络可视化》和《【GraphQuery】使用GraphQuery查询必应学术》
下面是一些比较经典的FBA计算分析的软件程序可以供大家进行选择:
生成FBA矩阵模型
在上一个步骤中,生成代谢网络之后,就可以产生FBA分析所需要的矩阵数据以及约束向量信息了。
执行线性规划计算
关于线性规划计算的原理,大家可以阅读博客文章《使用R#语言求解线性规划问题》,里面有比较详细的原理说明以及代码实现介绍。
评价结果
- 单细胞视角下的微生物基因组代谢酶嵌入分析 - 2026年2月25日
- 基因组代谢酶层级嵌入 - 2026年2月23日
- 基因组功能吉布斯LDA主题建模 - 2026年2月23日

One response
[…] 在之前所记录的《记一次浅尝辄止的FBA代谢网络分析》工作之中:对于FBA模型所需要的代谢网络的重建,我们会需要将目标基因组中的蛋白序列集合与从UniProt序列库中抽取出来的具有KO编号的参考序列库做比较。基于KO编号的注释结果得到对应的KEGG代谢反应结果数据的注释,从而重建出整个代谢网络。 […]