
估计阅读时长: 11 分钟https://github.com/rsharp-lang/ggplot 在生物信息学中的组学数据分析领域内,有一个非常常见的数据可视化图表:应用于可视化两两组别比对结果的火山图。在火山图之中,X坐标轴一般是log2FC,纵坐标Y轴,则一般是t检验的pvalue的-log10转换之后的值。由于fold change有大于1的值,A/B大于1,表示A的表达量高于B的表达量,反之小于一表示A的表达量低于B的表达量。这样子fold change经过log2转换之后,就会出现负数,散点一般呈轴对称分布在X=0的位置周围。这样子绘制出来的散点图就有点类似于火山喷发的样子了。 Order by Date Name Attachments a679af1eb9ffbfbad48c18d563ea51f3 • 45 kB • 856 click […]
估计阅读时长: 7 分钟https://github.com/rsharp-lang/ggplot 一张统计图形就是从数据到几何对象(geometric object, 缩写为geom, 包括点、线、条形等)的图形属性(aesthetic attributes, 缩写为aes, 包括颜色、形状、大小等)的一个映射。此外, 图形中还可能包含数据的统计变换(statistical transformation, 缩写为stats), 最后绘制在某个特定的坐标系(coordinate system, 缩写为coord)中, 而分面(facet, 指将绘图窗口划分为若干个子窗口)则可以用来生成数据中不同子集的图形。 […]
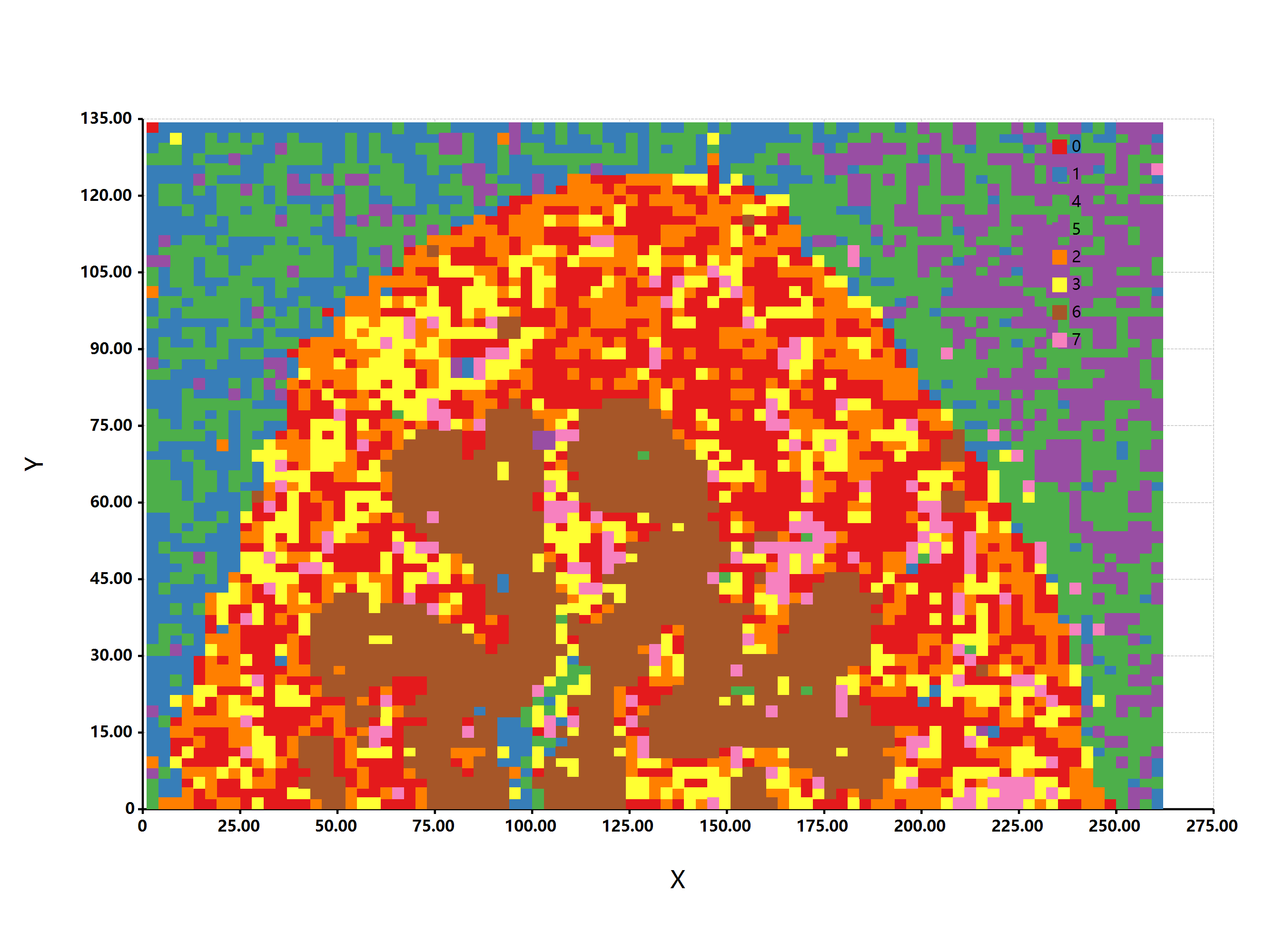
估计阅读时长: 15 分钟https://gcmodeller.org 在这篇博客文章之中,我主要是来详细介绍一下是如何从头开始实现Phenograph单细胞分型算法的。在之前的一篇博客文章《【单细胞组学】PhenoGraph单细胞分型》之中,我们介绍了Phenograph算法的简单原理,以及一个在R语言之中所实现的Phenograph算法的程序包Rphenograph。在这里我主要是详细介绍在GCModeller软件之中所实现的VisualBasic语言版本的Phenograph单细胞分型算法。 Attachments Rphenograph • 236 kB • 841 click 2021年9月20日
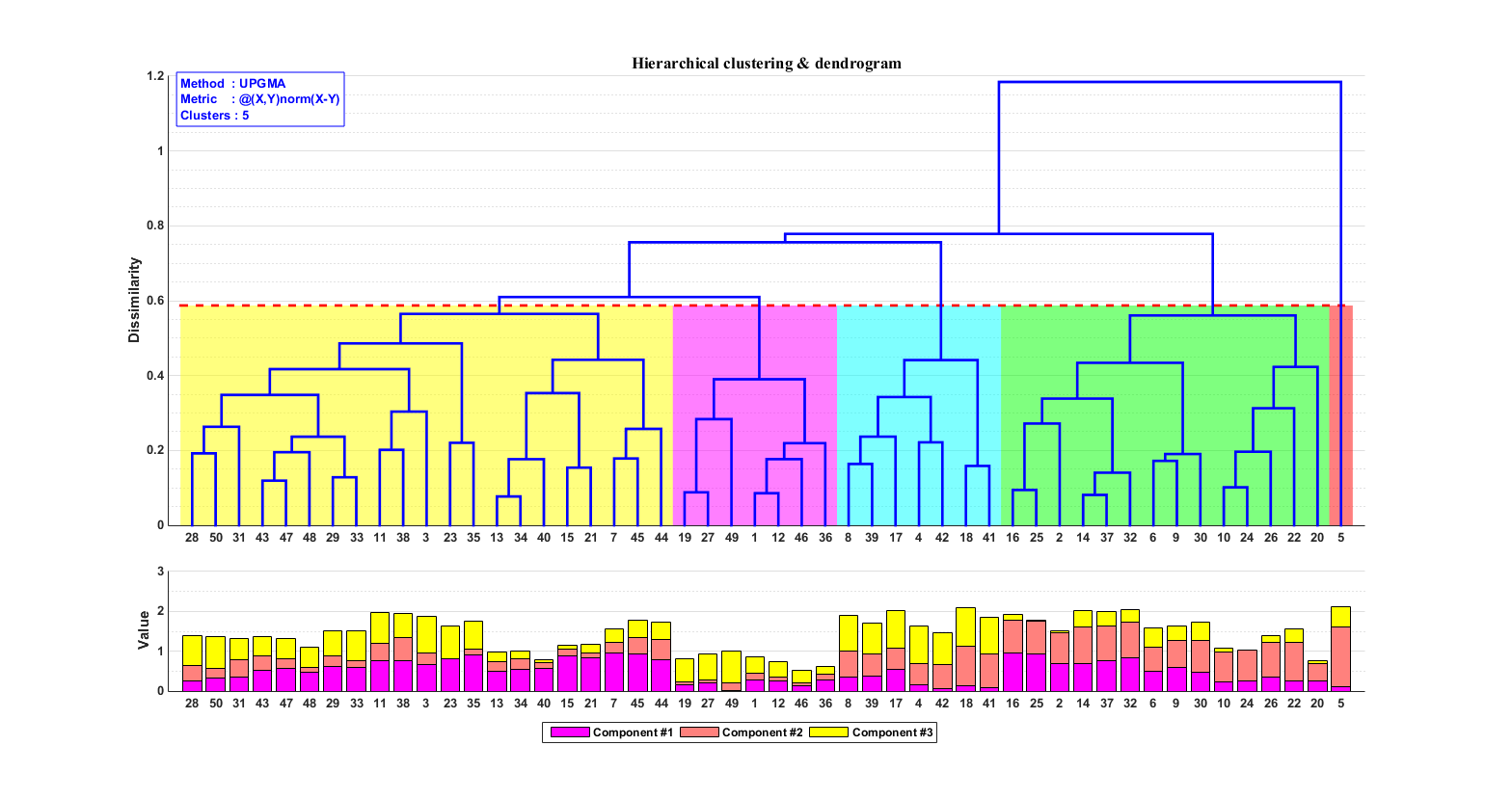
估计阅读时长: 14 分钟https://github.com/xieguigang/sciBASIC 层次聚类通过计算不同类别数据点间的相似度来创建一棵有层次的嵌套聚类树。基于层次聚类分析,我们可以初步可视化我们的一些原始数据: 例如对样本的层次聚类分类,可以让我们了解到样本在分组之间以及分组内的异质性。 对生物序列进行基于相似度的层次聚类分析,我们可以了解到序列之间的相似性程度或者进化关系 Order by Date Name Attachments metabolome • 14 kB • 867 click […]
估计阅读时长: 11 分钟PhenoGraph提供了与UMAP类似的算法过程进行单细胞组学数据的细胞分型处理操作。与UMAP方法相比,PhenoGraph并不会产生数据降维效果,仅仅产生数据点Cluster信息。如果需要将数据进行可视化,还需要借助于t-SNE算法将PhenoGraph的分型结果数据投影到一个二维平面上完成。 Order by Date Name Attachments Phenograph-image4 • 200 kB • 838 click 2021年8月9日Automated Optimal Parameters […]
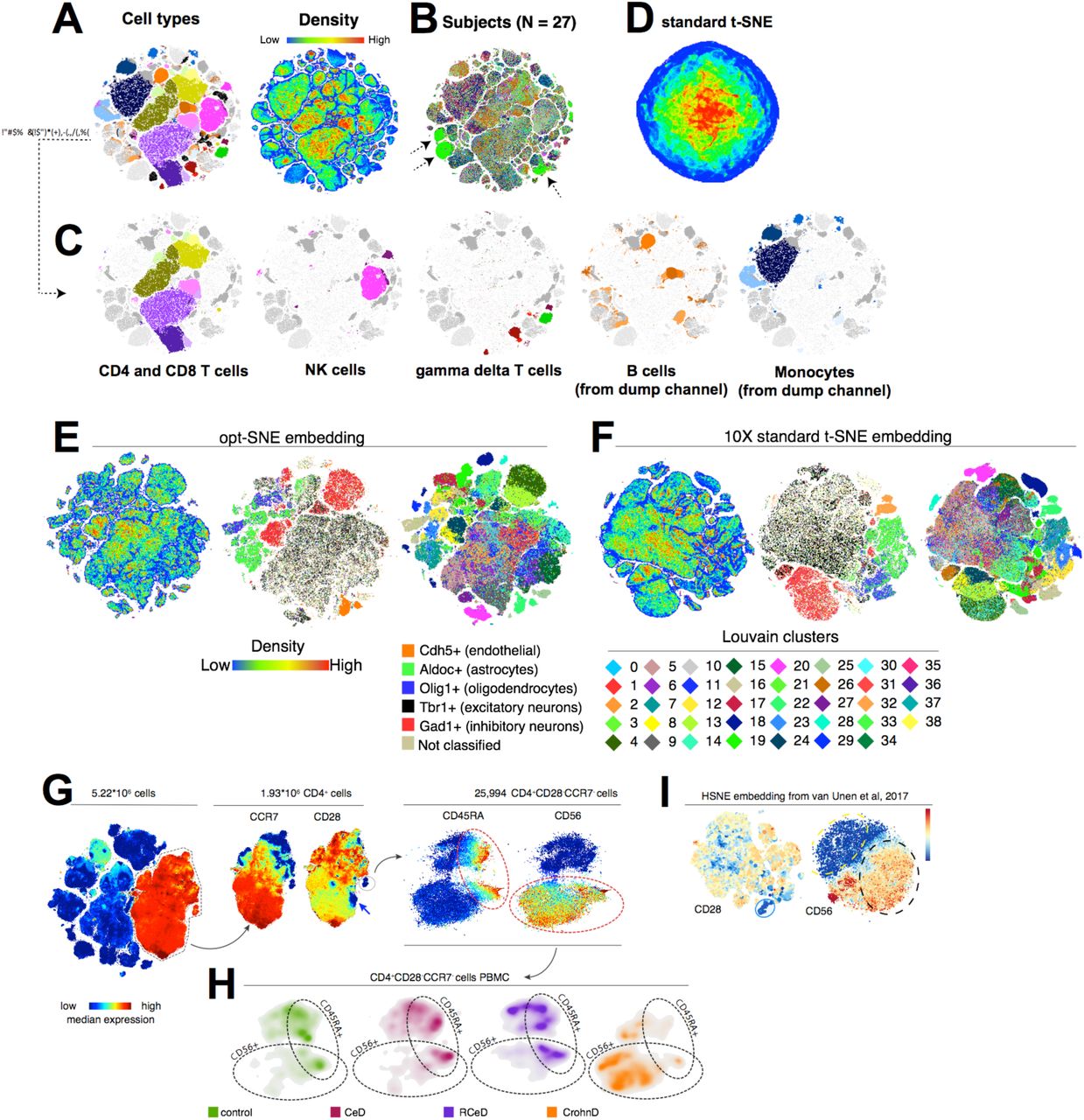
估计阅读时长: 16 分钟https://github.com/xieguigang/sciBASIC 等高线指的是地形图上高程相等的相邻各点所连成的闭合曲线。把地面上海拔高度相同的点连成的闭合曲线,并垂直投影到一个水平面上,并按比例缩绘在图纸上,就得到等高线。 Order by Date Name Attachments 1_Contour • 487 kB • 1010 click 2021年6月30日Ms1Contour • […]
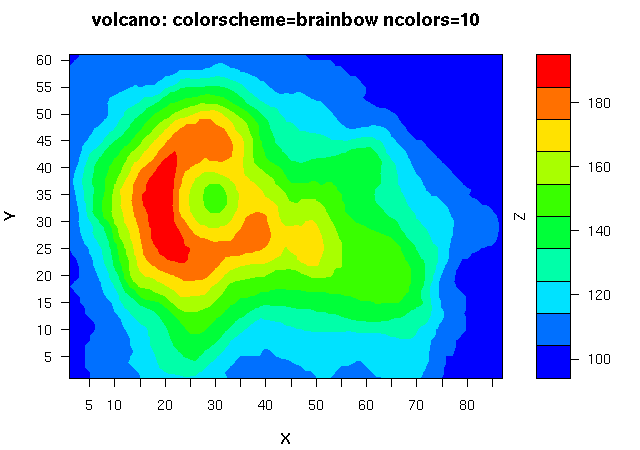
估计阅读时长: 23 分钟https://github.com/rsharp-lang/R-sharp 降维是将数据由高维约减到低维的过程而用来揭示数据的本质低维结构。它作为克服“维数灾难”的途径在这些相关领域中扮演着重要的角色。在过去的几十年里,有大量的降维方法被不断地提出并被深入研究,其中常用的包括传统的降维算法如PCA和MDS;流形学习算法如UMAP、t-SNE、ISOMAP、LE以及LTSA等。 Order by Date Name Attachments MNIST-LabelledVectorArray-60000x100 • 230 kB • 1011 click 2021年6月27日MNIST-LabelledVectorArray-60000x100Euclidean_Distance • […]
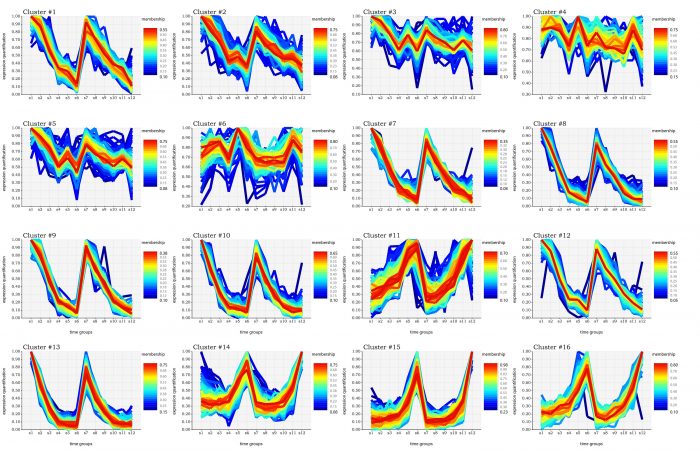
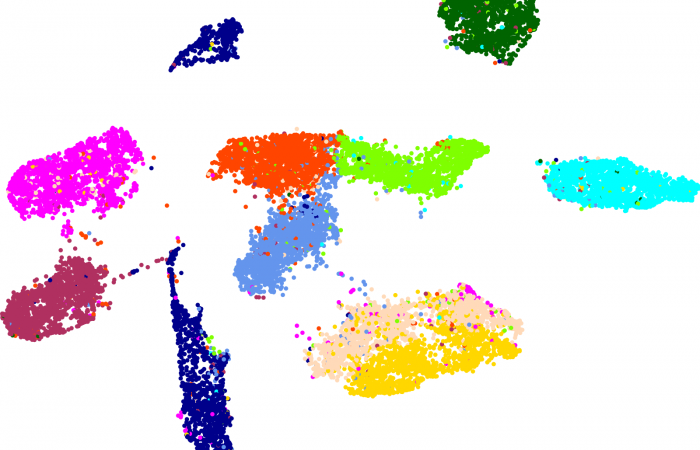
估计阅读时长: 11 分钟https://github.com/SMRUCC/GCModeller 在R语言之中,存在着一个用于进行表达数据的时间序列分析的程序包:TCseq。TCseq的全称为Time course sequencing,即时间序列分析,通过对表达矩阵进行时间上的模糊CMeans聚类,得到表达变化趋势一致的基因列表,进行基因表达的时间趋势分析。 在GCModeller之中,我仿照着TCseq程序包,自己编写了一个时间序列的聚类与可视化分析的R#程序包模块,在这里介绍给大家。 Order by Date Name Attachments Gene expression pattern visualization • 2 […]
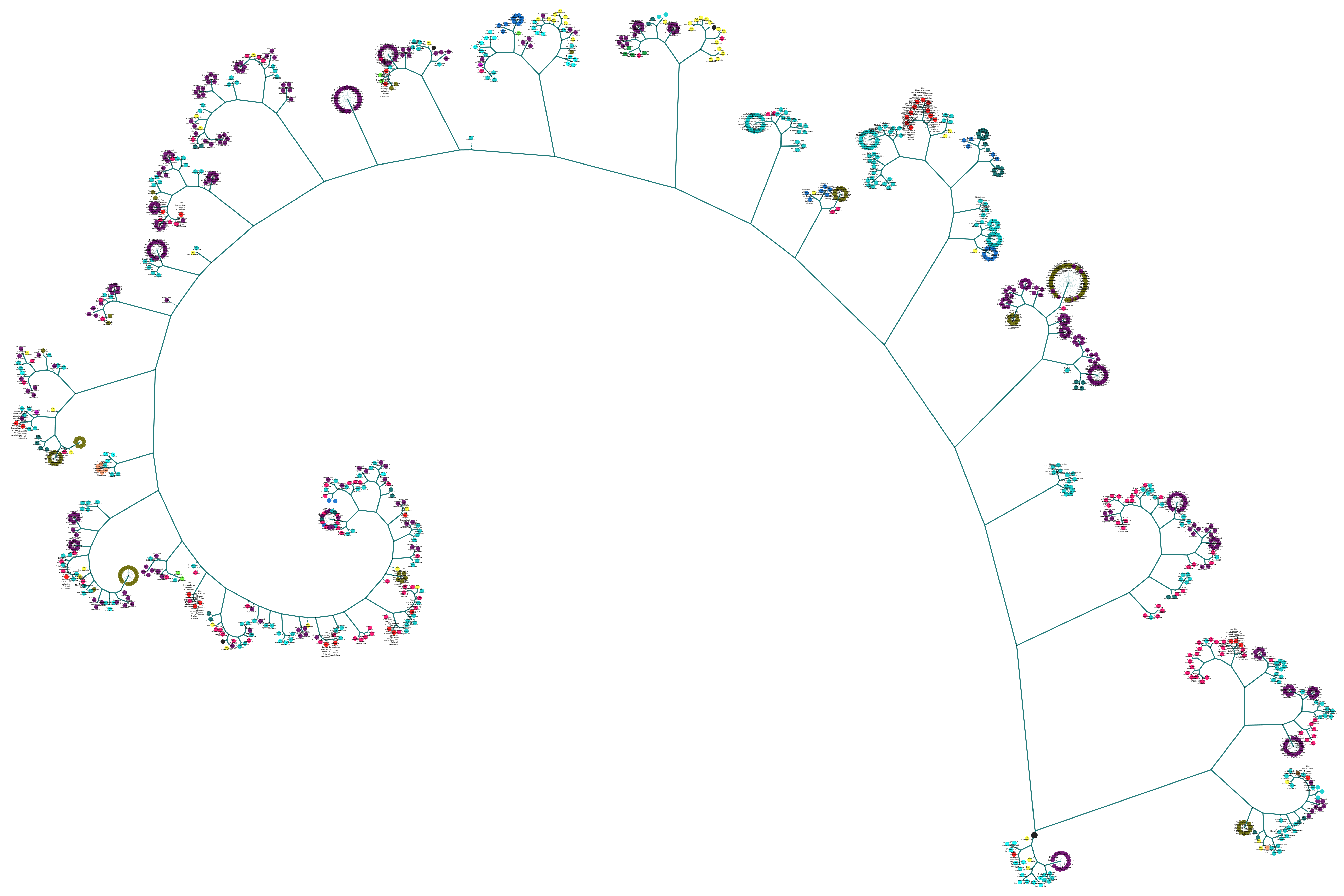
估计阅读时长: 4 分钟https://github.com/xieguigang/bclusterTree 对于二叉树,大家肯定不会陌生。二叉树其实就是一个有向无环图(有向:访问的方向是从父节点指向子节点;无环:子节点不会成为其父辈节点的父节点),大家可以从根节点一直往下访问到任意一个叶节点;节点间的方向是根据键值的比较的大小结果来建立的,大的值在右边,小的值在左边(《左迁与右迁》),零值在当前节点。 二叉树示意图来自于这篇博文《Self-balanced Binary Search Trees with AVL in JavaScript》 Order by Date Name Attachments Rplot […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?