
文章阅读目录大纲
 https://github.com/SMRUCC/GCModeller
https://github.com/SMRUCC/GCModeller
在R语言之中,存在着一个用于进行表达数据的时间序列分析的程序包:TCseq。TCseq的全称为Time course sequencing,即时间序列分析,通过对表达矩阵进行时间上的模糊CMeans聚类,得到表达变化趋势一致的基因列表,进行基因表达的时间趋势分析。
在GCModeller之中,我仿照着TCseq程序包,自己编写了一个时间序列的聚类与可视化分析的R#程序包模块,在这里介绍给大家。
![]()
模糊Cmeans聚类
我们在对表达量进行聚类分析的时候,一般可以使用Kmeans或者Cmeans这两种算法来完成。Cmeans与Kmeans算法的原理类似,都是基于欧几里得距离计算出对象与cluster之间的关联度的高低,从而完成聚类分析操作。从结果上来看,Cmeans与Kmeans理论上结果应该是相似的,而Cmeans方法相比较于Kmeans而言,其一个优势可能就是多出来了一个聚类的目标对象于对应的cluster之间的成员归属度这个结果值。
模糊c均值聚类融合了模糊理论的精髓。相较于k-means的硬聚类,模糊c提供了更加灵活的聚类结果。因为大部分情况下,数据集中的对象不能划分成为明显分离的簇,指派一个对象到一个特定的簇有些生硬,也可能会出错。故,对每个对象和每个簇赋予一个权值,指明对象属于该簇的程度。当然,基于概率的方法也可以给出这样的权值,但是有时候我们很难确定一个合适的统计模型,因此使用具有自然地、非概率特性的模糊c均值就是一个比较好的选择。

图片来自于 https://blog.csdn.net/lyxleft/article/details/88964494
在Cmeans聚类中。要达到的最终效果就是类内相似度最小,类间相似度最大,这个时候点和中心的加权距离之和就是最小的。所以Cmeans聚类最优解的表达式就是:

在上面的表达式中:
- μij指的就是隶属度值,元素j对类别i的隶属程度
- dij平方指的就是欧氏距离下元素j跟中心点i之间的距离,整个表示的就是各个点到各个类的加权距离的和。
- m是一个模糊化程度的参数。
我们可以将上面的J目标函数写为下面所示的VisualBasic代码:
<MethodImpl(MethodImplOptions.AggressiveInlining)>
Public Function J(m As Double, u As Double()(), centers As Double()(), entities As ClusterEntity()) As Double
Return centers _
.Select(Function(x, i)
Return entities _
.Select(Function(y, j1)
Return (u(j1)(i) ^ m) * Dist(y, x)
End Function) _
.Sum()
End Function) _
.Sum()
End Function对于成员隶属度u矩阵,我们可以通过下面的代码来完成计算:
Dim classIndex As Integer() = Enumerable.Range(0, classCount).ToArray
For i As Integer = 0 To u.Length - 1
Dim index As Integer = i
For j As Integer = 0 To u(i).Length - 1
Dim jIndex As Integer = j
Dim sumAll As Double = Aggregate x As Integer
In classIndex
Let d1 As Double = stdNum.Sqrt(Dist(entities(index), centers(jIndex)))
Let d2 As Double = stdNum.Sqrt(Dist(entities(index), centers(x)))
Let a As Double = d1 / d2
Let val As Double = a ^ (2 / (m - 1))
Into Sum(val)
u(i)(j) = 1 / sumAll
If Double.IsNaN(u(i)(j)) Then
u(i)(j) = 1
End If
Next
Next这个算法有一个约束条件,就是某一个元素对所有类别的隶属程度的值加起来要等于1。对于有约束条件的求极值问题,一般使用拉格朗日乘子法解决。我们构造出下面的拉格朗日函数,函数中共有三个变量,μij, di, 和λj,分别求偏导:

关于Cmeans的具体算法实现代码,大家可以阅读这个VisualBasic源代码文件:CMeans.vb。
While True
centers = GetCenters(classCount, fuzzification, u, entities, width).ToArray
j_new = J(fuzzification, u, centers, entities)
membership_diff = stdNum.Abs(j_new - j_old)
If j_old <> -1 AndAlso membership_diff < threshold Then
Exit While
Else
Call $"loop_{[loop]} membership_diff: |{j_new} - {j_old}| = {membership_diff}".__DEBUG_ECHO
End If
j_old = j_new
If parallel Then
Call u.updateMembershipParallel(entities, centers, classCount, fuzzification)
Else
Call u.updateMembership(entities, centers, classCount, fuzzification)
End If
If ++[loop] > maxLoop Then
Exit While
End If
End While从上面的源代码之中我们可以看到,我们要做的实际上就是不断的更新membership矩阵,使得J目标函数的值最小。当J函数的值小于给定的阈值之后,我们就可以结束计算了。
GCModeller中的基因表达趋势聚类分析
在GCModeller的geneExpression分析程序模块之中,提供了一个名称为expression.cmeans_pattern的函数进行TCseq类似的时间序列分析。这个函数的使用方法我们将在下面的小结中详细说明。
' Calculate gene expression pattern by cmeans algorithm.
'
' matrix: the gene expression matrix object which could be generated by loadExpression api.
' dim: the partition matrix size, it is recommended that width should be equals to the
' height of the partition matrix.
'
let expression.cmeans_pattern as function(matrix as <generic> Matrix,
dim as <generic> Object = "3,3",
fuzzification as double = 2,
threshold as double = 0.001) -> <generic> ExpressionPattern {
' .NET API information
' module: phenotype_kit.geneExpression
' LibPath: E:/GCModeller/GCModeller/bin/Library
' library: phenotype_kit.dll
' package: "geneExpression"
return call R#.interop_[geneExpression::CmeansPattern](...);
}R#代码示例
在GCModeller所提供的R#程序包之中,基因表达分析模块为phenotype_kit之中的geneExpression模块之中,所以我们首先需要导入geneExpression模块。因为对于这个模块而言,其不仅仅是用于处理时间序列,也可以用于处理普通的实验分组的结果数据。实际上时间序列就是一种变相的实验分组,只不过在时间序列分析之中,实验处理因子就是时间本身,每一个时间点就是一个处理组。所以在这里,我们除了导入了geneExpression模块之外,还导入了sampleInfo模块用于创建表达量数据预处理所需要使用到的实验分组信息。
在这里我使用TCseq程序包之中所提供的一个测试数据为大家演示在GCModeller之中进行基因表达趋势聚类分析的脚本。这个测试用的数据表格大家也可以在R环境之中,读取TCseq程序包的countsTable数据集:
data("countsTable");
head(countsTable);
# s1 s2 s3 s4 s5 s6 s7 s8 s9 s10 s11 s12
# peak1 344 243 169 70 57 20 298 199 135 63 54 34
# peak2 72 114 91 93 133 164 55 71 93 116 150 191
# peak3 28 50 109 115 94 109 60 89 75 129 85 101
# peak4 28 49 110 113 103 108 59 89 77 131 83 104
# peak5 464 344 280 154 108 33 444 255 259 155 97 32
# peak6 175 129 77 29 15 4 134 91 44 33 11 5在脚本之中,我们会需要首先使用read.csv函数直接从github的代码库之中读取这个demo用的表格文件。在geneExpression程序模块之中,提供了一些进行表达量原始数据处理相关的函数,例如:
- load.expr,这个函数接受一个文件对象或者数据框,加载为基因表达数据对象
- average,这个函数在基因表达数据对象和给定的实验分组信息的基础上,计算出每一个基因在不同的实验分组之中的表达的平均值。(如果计算foldchange,会需要这个平均值结果)
- relative,这个函数对基因表达数据对象进行数据缩放,这个函数只是进行简单的
x/max(x) - expression.cmeans_pattern,随后呢,就可以使用这个函数进行基因表达数据对象的趋势聚类分析了
根据expression.cmeans_pattern的名字就可以看得出来,在这里是使用Cmean模糊聚类进行表达趋势的聚类分析计算的。在使用这个函数计算出表达量趋势的聚类结果后,就可以通过plot函数可视化出来了。
下面我给出了简单的使用上面的时间序列分析模块进行基因表达数据的时间趋势聚类分析以及对应的结果数据可视化的R#脚本代码。
imports "visualPlot" from "visualkit";
imports ["geneExpression", "sampleInfo"] from "phenotype_kit";
bitmap(file = `${dirname(@script)}/patterns.png`) {
const patterns = "github://SMRUCC/GCModeller/master/src/workbench/R%23/demo/HTS/counts.csv"
|> read.csv(row_names = 1)
|> load.expr(rm_ZERO = TRUE)
|> average(sampleinfo = sampleInfo(
ID = ["s1", "s2", "s3", "s4", "s5", "s6", "s7", "s8", "s9", "s10", "s11", "s12"],
sample_name = ["s1", "s2", "s3", "s4", "s5", "s6", "s7", "s8", "s9", "s10", "s11", "s12"],
sample_info = ["s1", "s2", "s3", "s4", "s5", "s6", "s7", "s8", "s9", "s10", "s11", "s12"]
))
|> relative
|> expression.cmeans_pattern(
dim = [4, 4],
fuzzification = 2,
threshold = 0.005
)
;
print("view patterns result:");
print(patterns);
plot(patterns,
size = [9000, 6000],
colorSet = "Jet",
axis_label.cex = "font-style: normal; font-size: 14; font-family: Microsoft YaHei;"
);
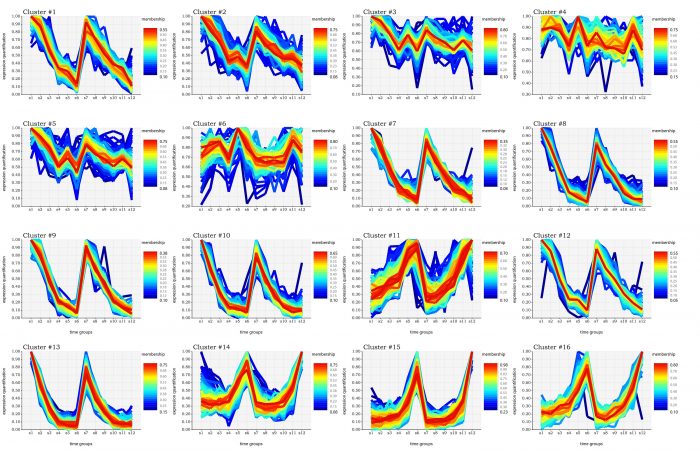
}我们将上面的demo测试代码保存为脚本文件,然后在命令行之中使用R#命令执行,产生的基因表达时间趋势聚类结果的可视化图如下:

在上面的结果图之中,一个cluster就是对一群表达量变化趋势很相似的基因的集合的在时间上的表达量可视化。上面的可视化图之中,X坐标轴为时间序列或者我们的实验分组,Y坐标轴则是进行数据缩放后得到的相对的表达量值。在每一个图标之中,我们对每一个基因的表达量都绘制了一条曲线,并且用不同的颜色对这个基因与当前的cmeans聚类结果的归属度进行了显示。曲线的颜色偏红色表示基因与当前的聚类的成员归属度越高,反之蓝色表示归属度越低。
理论上,我们可以修改expression.cmeans_pattern函数的dim维数参数来产生任意数量的聚类结果。除了修改聚类结果数量外,我们还可以通过修改plot函数的参数值来进行一些样式的调整,例如调整颜色谱为ColorBrewer颜色谱之中的BuPu颜色列表:

- 单细胞视角下的微生物基因组代谢酶嵌入分析 - 2026年2月25日
- 基因组代谢酶层级嵌入 - 2026年2月23日
- 基因组功能吉布斯LDA主题建模 - 2026年2月23日

One response
[…] C均值聚类 […]