
文章阅读目录大纲
最近的工作中我需要按照之前的这篇博客文章《基因组功能注释(EC Number)的向量化嵌入》中所描述的流程,将好几十万个微生物基因组的功能蛋白进行酶编号的比对注释,然后基于注释结果进行向量化嵌入然后进行数据可视化。通过R#脚本对这些微生物基因组的蛋白fasta序列的提取操作,最终得到了一个大约是58GB的蛋白序列。然后将这个比较大型的蛋白序列比对到自己所收集到的ec number注释的蛋白序列参考数据库之上。
很显然,假若我仍然是直接通过之前的流程终所使用ncbi blastp工具做比对,整个计算过程中,光blastp同源收缩注释这个过程所花费的时间很可能是好几个星期的时间,这个是无法被接受的。但是好在通过查询文献,发现了一款号称比ncbi的blast+软件的运行效率高了好几千倍的序列搜索工具:diamond软件。Diamond 是一款专为蛋白质序列比对设计的高性能工具,其速度远超传统的 BLAST+ 工具集。在敏感度相当的情况下,Diamond 可以比标准 BLASTP 快数个数量级。这种卓越的速度使 Diamond 能够在数小时内完成原本需要数月的超大规模比对任务。
下面的是我的工作中所需要处理的数据集:
(base) zysw@zysw-MZ72-HB2-00:~/sdb/xieguigang/20260213$ ls -lh
总计 450G
-rw-r--r-- 1 root root 505M 2月 13 18:00 ec_numbers.dmnd
-rwxr--r-- 1 root root 491M 2月 13 17:58 ec_numbers.fasta
-rw-rw-r-- 1 zysw zysw 19G 2月 15 05:51 ec_numbers_results.tar.gz
-rw-r--r-- 1 root root 372G 2月 14 18:32 ec_numbers_results.tsv
-rw-rw-r-- 1 zysw zysw 58G 2月 13 06:06 prot_huge.fasta同样的,我们需要进行注释的蛋白序列数据集也是同样的下面所示的fasta标题格式的序列文件:
现在,基于上面所提到的所需要进行处理的数据集,我将使用diamond软件处理上面的blastp序列比对的整个过程记录了下来。
Diamond软件使用教程
Diamond blastp搜索的步骤
假若你的linux服务器环境中已经安装有conda环境的化,可以非常简单的直接按照下面的命令行进行安装工作:
conda install -c bioconda diamond按照上面的命令行在conda环境中安装好diamond软件后,就可以直接使用了。与ncbi的blast+程序一样,进行基于diamond软件的blastp搜索注释同样也需要首先建立起参考数据库,然后才能够进行后续的blastp搜索注释操作。首先第一步,建立参考数据库索引,可以通过如下命令行参数来实现:
diamond makedb --in ec_numbers.fasta -d ec_numbers第二步,进行blastp搜索注释。由于diamond软件在默认情况下,会按照query序列的fasta标题的第一个空格进行标题的截断,由于我的fasta序列数据中,在标题上会包含有蛋白的编号以及对应的物种来源信息。直接按照默认的格式进行输出,会将序列标题中的物种信息丢失,导致后续的数据处理额外增加一个物种信息匹配的过程。所以在这里做blastp搜索注释,我直接在命令行参数中设置了qtitle和stitle这两个标题信息来直接输出原始的标题字符串,来避免默认情况下的标题信息丢失的问题。
diamond blastp -d ec_numbers -q prot_huge.fasta -o ec_numbers_results.tsv \
--very-sensitive --threads 1024 \
--outfmt 6 qtitle stitle pident length mismatch gapopen qstart qend sstart send evalue bitscore按照上面的命令行设置,大约比对了一个小时左右,就完成了我的蛋白序列注释的工作。这个计算效率比ncbi的blast+相比较,简直是一个天一个地的区别。
Diamond 的基本用法与 BLAST+ 类似,需要指定数据库、查询序列和输出文件等基本参数。在这里我们所使用到的基本参数如下:
- 数据库参数 --db (-d):指定 Diamond 数据库文件路径(包括文件名但不包含扩展名)。例如 -d nr 表示使用当前目录下的 nr.dmnd 数据库。
- 查询参数 --query (-q):指定输入查询序列文件,支持 FASTA 或 FASTQ 格式,可以是 gzip 压缩文件。例如 -q queries.faa 指定查询文件为 queries.faa。
- 输出参数 --out (-o):指定比对结果输出文件名。例如 -o output.txt 将结果输出到 output.txt 文件中。
- 线程参数 --threads (-p):设置并行运行的线程数。默认情况下 Diamond 会自动检测并使用所有可用的 CPU 核心。在集群环境下,可根据分配的计算资源设置线程数,以避免超负荷运行。
完成diamond blastp搜素程序的运行后,会生成类似于下面的截图中所展示结果表格文件:

Diamond 与 BLAST+ 在功能和用法上高度兼容,可作为 NCBI BLAST 的直接替代工具。它支持 BLASTP 和 BLASTX 两种主要比对模式,分别用于蛋白质查询序列对蛋白质数据库的搜索,以及核酸查询序列翻译后对蛋白质数据库的搜索。Diamond 的输入输出格式与 BLAST+ 基本一致,支持标准的 FASTA/FASTQ 查询文件和 BLAST 格式的数据库。输出结果默认采用 BLAST 制表格式 (format 6),方便后续脚本处理和解析。这种兼容性使得研究人员可以无缝地将 Diamond 集成到现有的分析流程中,而无需大幅修改脚本或参数设置。
虽然说使用Diamond软件可以很好的兼容目前的ncbi blast+流程,但是针对diamond软件的结果输出的表格格式我有一个比较想吐槽的点就是,他的表格结果输出是没有标题行的。也就是我们在拿到一个diamond软件的结果表格之后,假若我们并不是进行blastp比对操作的实际操作者,在这种情况下,由于我们并不太清楚运行diamond软件的时候在命令行参数中对结果表格格式的具体设置的参数内容,导致我们在这种情况下就只能够靠自己的推测来进行表格结果数据的解析。
Diamond灵敏度模式全谱系(从快到慢,从宽松到严格)
假若我们在diamond软件做blastp搜索的时候,不设定任何灵敏度模式的话,diamond软件会非常快速的大致搜索出一些同源性很高的序列结果。但是可能会遗漏掉一些较低同源性的序列结果。假若我们想要自主设定diamond软件做blastp搜索的灵敏度,可以参考下面的表格来设置。
| 模式 | 速度 | 灵敏度 | 适用场景 |
|---|---|---|---|
| 默认模式(无参数) | ⚡⚡⚡ 最快 | 最低 | 寻找高同一性(>70%)的快速匹配 |
| --fast | ⚡⚡ 快 | 较低 | 速度优先的大规模筛查 |
| --more-sensitive | ⚡⚡ 中快 | 中等 | 平衡模式,一般用途 |
| --sensitive | ⚡ 中慢 | 中高 | 寻找中等同源性(>40%) |
| --very-sensitive | 🐢 慢 | 高 | 综合最佳选择,适合大多数科研分析 |
| --ultra-sensitive | 🐢⚡ 最慢 | 最高 | 寻找极远缘同源序列,计算成本最高 |
按照上面的参数和场景,我们可以绘制下面的流程来决定diamond做blastp搜索的时候的灵敏度的参数设置:
需要找远缘同源物?-→ 是 -→ 计算资源充足?-→ 是 -→ 使用`--ultra-sensitive`
↓ ↓
否 否
↓ ↓
┌───────┴───────────┐ 使用`--very-sensitive`
↓ ↓
时间紧迫? 数据库质量高?
| ↓ ↓ ↓
| 是 -→ 用 `--sensitive` 否 是 -→ 用`--very-sensitive`
↓ ↓
否 -→ 用`--very-sensitive` 用`--sensitive`其他结果过滤参数
Diamond 还提供了多种参数用于过滤比对结果,以确保输出结果的可靠性:
- 期望值阈值 --evalue (-e):设置报告比对结果的最大期望值 (E-value)。E-value 越小表示匹配的统计显著性越高。默认情况下 Diamond 使用 0.001 作为阈值,即只报告 E-value 小于 0.001 的比对。这个阈值比传统 BLAST 的默认值 10 更严格,旨在过滤掉大量低质量的随机匹配。用户可根据需要调整阈值,例如使用更严格的 -e 1e-5 或宽松的 -e 10。
- 最小比特得分 --min-score:设置报告比对结果的最小比特得分。此参数可替代 E-value 阈值,用于过滤低质量的比对。通常情况下,使用 E-value 阈值即可满足需求。
- 序列一致性阈值 --id:只报告序列一致性百分比高于给定值的比对。例如 --id 50 只输出一致性 >50% 的匹配。这在需要过滤掉低相似度匹配时有用,但需注意序列一致性受比对长度影响,应结合覆盖率等指标一起考虑。
- 查询覆盖率阈值 --query-cover:只报告查询序列覆盖率高于给定值的比对。覆盖率指查询序列参与比对部分的长度占查询序列总长的比例。此参数可用于过滤掉仅部分匹配的结果,例如要求查询序列至少有 80% 的长度参与比对,可设置 --query-cover 80。
- 最大目标序列数 --max-target-seqs (-k):限制每个查询序列报告的匹配序列数量。默认情况下 Diamond 不限制匹配数,可能为每个查询输出大量结果。使用 -k 参数可以只保留得分最高的若干个匹配。例如 -k 1 表示只输出每个查询序列的最佳匹配。这在功能注释时常用,以避免后续处理过多的冗余结果。
- 最佳匹配百分比范围 --top:报告相对于最佳匹配得分一定百分比范围内的所有比对。例如 --top 10 将报告所有得分不低于最佳得分 90% 的匹配。该参数可用于获取每个查询序列的近似最优匹配集合,常用于去除冗余匹配或保留多个可能的功能注释候选。
以上筛选参数可以组合使用,以满足不同的分析需求。例如,在进行蛋白质功能注释时,可以考虑使用严格的 E-value 阈值并结合 --top 参数,以获得每个查询序列的可靠匹配集合。而在进行同源基因聚类分析时,可能需要放宽阈值并保留多个匹配,此时可适当提高 --evalue 并设置较大的 --max-target-seqs 或不限制。
处理Diamond搜索注释结果
通过diamond软件完成了上面的blastp搜索注释之后,生成了一个大约是371GB的表格文件。很显然我们没有办法按照之前的处理流程来走一遍这个数据。如此大的数据量,在个人电脑上做处理,内存会裂开。
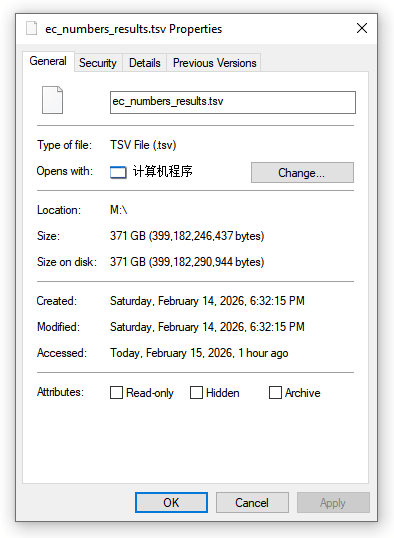
在cmd上通过more命令轻轻的peeks了一下这个大表格文件的前几行数据,通过观察发现,diamond软件虽然在进行搜索注释的时候是并行化计算的。但是结果表格中所输出的结果数据是顺序排列的。这个就意味着我们可以按照流式方式处理这个表格文件中的数据分组。按照我们做diamond软件做搜索注释的时候所设定的结果表格输出的格式参数,我们可以设计出如下所示的表格对象来读取上面的结果表格的内容:
Imports System.Runtime.CompilerServices
Imports Microsoft.VisualBasic.ComponentModel.Map(Of String, String)
Imports SMRUCC.genomics.ComponentModel.Annotation
Imports SMRUCC.genomics.Interops.NCBI.Extensions.LocalBLAST.Application.BBH
''' <summary>
''' 代表 DIAMOND BLASTP 结果文件 (.m8) 中的一行记录
''' </summary>
Public Class DiamondAnnotation : Implements IBlastHit, IMap
''' <summary>
''' 1. 查询序列ID
''' </summary>
''' <returns></returns>
Public Property QseqId As String Implements IBlastHit.queryName, IMap.Key
''' <summary>
''' 2. 目标序列ID
''' </summary>
''' <returns></returns>
Public Property SseqId As String Implements IBlastHit.hitName, IBlastHit.description, IMap.Maps
''' <summary>
''' 3. 比对一致性百分比
''' </summary>
''' <returns></returns>
Public Property Pident As Double
''' <summary>
''' 4. 比对长度
''' </summary>
''' <returns></returns>
Public Property Length As Integer
''' <summary>
''' 5. 错配数
''' </summary>
''' <returns></returns>
Public Property Mismatch As Integer
''' <summary>
''' 6. Gap 打开次数
''' </summary>
''' <returns></returns>
Public Property GapOpen As Integer
''' <summary>
''' 7. 查询序列起始位置
''' </summary>
''' <returns></returns>
Public Property QStart As Integer
''' <summary>
''' 8. 查询序列结束位置
''' </summary>
''' <returns></returns>
Public Property QEnd As Integer
''' <summary>
''' 9. 目标序列起始位置
''' </summary>
''' <returns></returns>
Public Property SStart As Integer
''' <summary>
''' 10. 目标序列结束位置
''' </summary>
''' <returns></returns>
Public Property SEnd As Integer
''' <summary>
''' 11. E-value (期望值)
''' </summary>
''' <returns></returns>
Public Property EValue As Double
''' <summary>
''' 12. Bit Score (比特得分)
''' </summary>
''' <returns></returns>
Public Property BitScore As Double
Public Overrides Function ToString() As String
Return $"{QseqId} vs {SseqId} | Identity: {Pident}% | E-value: {EValue}"
End Function
End Class上面的数据结构实际上就是定义了NCBI BLAST 的制表格式结果输出格式 (format 6)。这样子,我们就可以通过下面的流式数据处理代码来完成这个371GB的表格数据的解析处理:
Private Shared Iterator Function SimpleStreamLoader(filepath As String) As IEnumerable(Of String)
Dim line As Value(Of String) = ""
Using reader As New StreamReader(filepath)
' 逐行读取
While (line = reader.ReadLine()) IsNot Nothing
' 去除首尾空白
line = line.Trim()
' 跳过空行
If String.IsNullOrEmpty(line) Then
Continue While
' 跳过注释行 (以 # 开头)
ElseIf line.StartsWith("#") Then
Continue While
End If
Yield line
End While
End Using
End Function
Private Shared Function ParseLine(line As String, lineNum As Integer) As DiamondAnnotation
' 按制表符分割列
Dim parts As String() = line.Split(ControlChars.Tab)
' .m8 标准格式应有 12 列
If parts.Length <> 12 Then
' 可以在这里记录日志,或者直接跳过格式错误的行
Call $"第 {lineNum} 行格式错误 (列数: {parts.Length}),已跳过。".warning
Return Nothing
End If
' 创建新对象并赋值
Dim hit As New DiamondAnnotation()
hit.QseqId = parts(0)
hit.SseqId = parts(1)
' 使用 InvariantCulture 解析数字,以确保能正确处理小数点 "."
' 以及科学计数法 (例如 2.31e-84)
hit.Pident = Double.Parse(parts(2), CultureInfo.InvariantCulture)
hit.Length = Integer.Parse(parts(3), CultureInfo.InvariantCulture)
hit.Mismatch = Integer.Parse(parts(4), CultureInfo.InvariantCulture)
hit.GapOpen = Integer.Parse(parts(5), CultureInfo.InvariantCulture)
hit.QStart = Integer.Parse(parts(6), CultureInfo.InvariantCulture)
hit.QEnd = Integer.Parse(parts(7), CultureInfo.InvariantCulture)
hit.SStart = Integer.Parse(parts(8), CultureInfo.InvariantCulture)
hit.SEnd = Integer.Parse(parts(9), CultureInfo.InvariantCulture)
hit.EValue = Double.Parse(parts(10), CultureInfo.InvariantCulture)
hit.BitScore = Double.Parse(parts(11), CultureInfo.InvariantCulture)
Return hit
End Function在上面的流式数据处理代码中,对于大型的文本数据集合,会默认显示出一个进度条来显示目标大型文本文件的处理进度。程序会一行一行的从目标文件中读取文本数据出来,然后再按照TAB符号进行行的切割,然后填充到对应的属性值上,完成整个文本结果文件的解析操作。基于上面的diamond blastp比对结果,我们得到的是单个方向的比对注释结果。
在前面的流程中,根据单个方向的搜索注释结果,结果可能没有双向比对结果的准确度高,但是按照解析出来的hits中的term id的supports降序排序的方式进行单向比对注释,仍然可以得到一定比较高准确度的注释结果,基于此,我们可以构建出下面的数据结构来进行单向分组结果数据的存储:
Public Class RankTerm : Implements INamedValue
Public Property queryName As String Implements INamedValue.Key
Public Property term As String
Public Property source As String()
Public Property scores As Double()
''' <summary>
''' SUM of the all scores for this term
''' </summary>
''' <returns></returns>
Public ReadOnly Property score As Double
Get
Return scores.SafeQuery.Sum
End Get
End Property
Public ReadOnly Property supports As Integer
Get
Return source.TryCount
End Get
End Property
Public ReadOnly Property topHit As String
Get
If scores.IsNullOrEmpty OrElse source.IsNullOrEmpty Then
Return Nothing
End If
Return source(which.Max(scores))
End Get
End Property
Public Overrides Function ToString() As String
Return $"[{term}] {queryName} = {score}"
End Function
End Class在下面的代码中,由于我们假设diamond会按照顺序进行比对结果的输出,所以我们假设下面的代码会正常的按照diamond软件所输出的hit结果中的query信息,进行分组。得到了query的分组之后,就可以根据结果term的supports结果来完成对应的ec number的简单注释操作:
Imports System.Runtime.CompilerServices
Imports Microsoft.VisualBasic.ComponentModel.DataSourceModel
Imports Microsoft.VisualBasic.Linq
Imports SMRUCC.genomics.Interops.NCBI.Extensions.Pipeline
''' <summary>
''' Processing term assignment via stream processing of the large diamond annotation result output
''' </summary>
''' <param name="m8"></param>
''' <returns></returns>
<Extension>
Public Iterator Function MakeTerms(m8 As IEnumerable(Of DiamondAnnotation), Optional topBest As Boolean = True) As IEnumerable(Of RankTerm)
Dim group As New List(Of DiamondAnnotation)
Dim query As String = ""
Dim rank As Func(Of DiamondAnnotation, Double) = Function(hit) (hit.Pident + 1) * (hit.BitScore + 1)
For Each hit As DiamondAnnotation In m8.SafeQuery
If hit.QseqId <> query Then
If group.Count > 0 Then
Dim data As New NamedCollection(Of DiamondAnnotation)(query, group)
Dim terms = RankTerm.MeasureTopTerm(data, rank, Nothing)
Call group.Clear()
If topBest AndAlso terms.Any Then
Yield terms.First
Else
For Each term As RankTerm In terms
Yield term
Next
End If
End If
query = hit.QseqId
End If
Call group.Add(hit)
Next
End Function之后,再按照query中的genbank的id进行基因组来源分组,构建出基因组的注释结果内容。再下面的所示的两段R#脚本实现了上面所描述的流式数据处理流程:
require(GCModeller);
imports ["annotation.workflow", "annotation.terms"] from "seqtoolkit";
let rawdata = read_m8(relative_work("ec_numbers_results.tsv"), stream = TRUE);
let stream = open.stream(relative_work("ec_terms.csv"),type = "terms");
stream.flush(m8_metabolic_terms(rawdata), stream);上面的脚本大概会花费掉3个半小时处理完整个300多GB的diamond结果输出表格,最终得到了约38GB的酶注释结果表格。然后我们就可以再基于下面的一段R#脚本完成从单个基因的酶注释结果合并输出为基因组的代谢酶注释结果,大约花费半个小时处理完这差不多40GB的表格文件为注释结果json文件:
require(GCModeller);
imports ["annotation.workflow", "annotation.terms"] from "seqtoolkit";
let stream = open.stream(relative_work("ec_terms.csv"),type = "terms", ioRead = TRUE);
let genomes = make_vectors(stream, stream = TRUE);
write_genomes_jsonl(genomes, file = relative_work("genomes_metabolic.jsonl"));数据可视化
我们在这里仍然是将按照TF-IDF算法进行向量化嵌入后的结果,进行UMAP降维,然后进行降维后的散点图可视化的操作来对我们的结果进行绘图可视化:
require(GCModeller);
require(umap);
imports "taxonomy_kit" from "metagenomics_kit";
let models = read.jsonl(file = relative_work("genomes_metabolic.jsonl"), what = "genome_vector");
let vec = models |> tfidf_vectorizer();
write.csv(vec, file = relative_work("metabolic_embedding.csv"));
let data = vec |> select(-1) |> set_rownames(vec$taxonomy);
let result = umap(data, neighbors = 128,
local_connectivity = 2,
method = "Cosine");
let manifold = as.data.frame(result$umap, labels = result$labels);
let taxon = biom_string.parse(rownames(manifold));
manifold[,"phylum"] <- taxonomy_name(taxon, rank = "phylum");
manifold[,"class"] <- taxonomy_name(taxon, rank = "class");
manifold[,"order"] <- taxonomy_name(taxon, rank = "order");
write.csv(result, file = "umap.csv");
通过散点图可视化可以发现,在通过diamond软件来处理更加大型的数据集的blastp搜索注释下,我们得到的基因组的嵌入结果可以很好的对基因组进行物种分类工作。在我们所得到的基因组向量化嵌入结果的散点图上,相同颜色的散点(相同Phylum分类下的基因组)都是相互聚集在一起,说明我们的基于diamond搜索外加向量嵌入得到的基因组代谢功能注释向量化嵌入结果可以很好的提取出基因组的代谢特征。基于这些得到的特征向量结果值,我们可以很方便的用于下游的机器学习建模之类的工作
参考文献
- Buchfink B, Reuter K, Drost HG. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 2021 Apr;18(4):366-368. doi: 10.1038/s41592-021-01101-x. Epub 2021 Apr 7. PMID: 33828273; PMCID: PMC8026399.
- 单细胞视角下的微生物基因组代谢酶嵌入分析 - 2026年2月25日
- 基因组代谢酶层级嵌入 - 2026年2月23日
- 基因组功能吉布斯LDA主题建模 - 2026年2月23日

3 Responses
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
谢老师,写快点呀,在看着你更新文章呢。
😲啊?