

估计阅读时长: 8 分钟为了能够稳定的使用Ubuntu系统作为我的测试环境,我专门安装了Ubuntu的2404 LTS版本。但是发现虽然是LTS版本,仍然会因为系统更新而出现各种莫名奇妙的BUG。例如,在今天我为了安装软件,使用apt upgrade更新系统环境之后,出现了下面的消息: Order by Date Name Attachments ubuntu-24-04-noble-numbat • 66 kB • 537 click 2025年6月7日ti8S1 […]

估计阅读时长: 6 分钟CentOS查看系统版本信息 cat /etc/redhat-release # CentOS Stream release 8 cat /proc/version # Linux version 4.18.0-489.el8.x86_64 (mockbuild@x86-05.stream.rdu2.redhat.com) (gcc […]
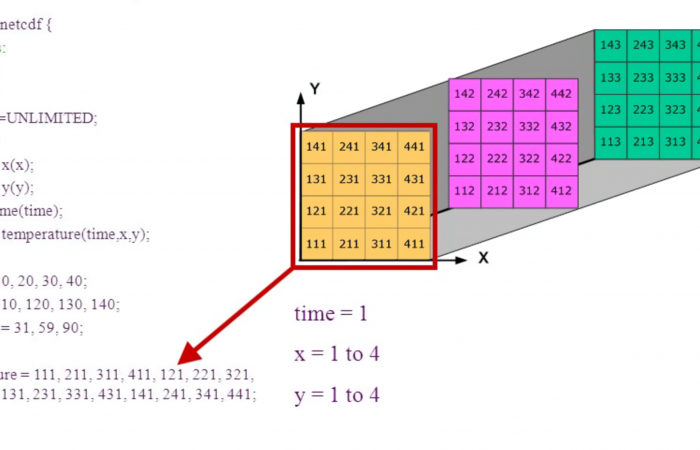
估计阅读时长: 15 分钟https://github.com/xieguigang/Darwinism NetCDF文件格式(Network Common Data Format)是一种以network byteorder进行编码的CDF数据文件格式。其广泛应用于大气科学、水文、海洋学、环境模拟、地球物理等诸多数据科学计算分析领域内的数据存储。 Order by Date Name Attachments netcdf • 2 MB • […]

估计阅读时长: 8 分钟https://github.com/xieguigang/Darwinism 对于LINQ数据查询引擎而言,其可以接收任意类型的数据源,进行数据查询。只要存在有相对应的数据源驱动程序即可。 Order by Date Name Attachments sqlite • 18 kB • 798 click 2021年6月19日sqlite-contents • […]

估计阅读时长: 11 分钟https://github.com/xieguigang/Darwinism LINQ(Language Integrated Query)技术是一种语言集成查询,即LINQ是VisualBasic语言之中的一种语法。其由微软公司于.NET Framework 3.5引入的一种SQL查询语言非常相似的数据查询语法。 Order by Date Name Attachments query • 51 kB • […]
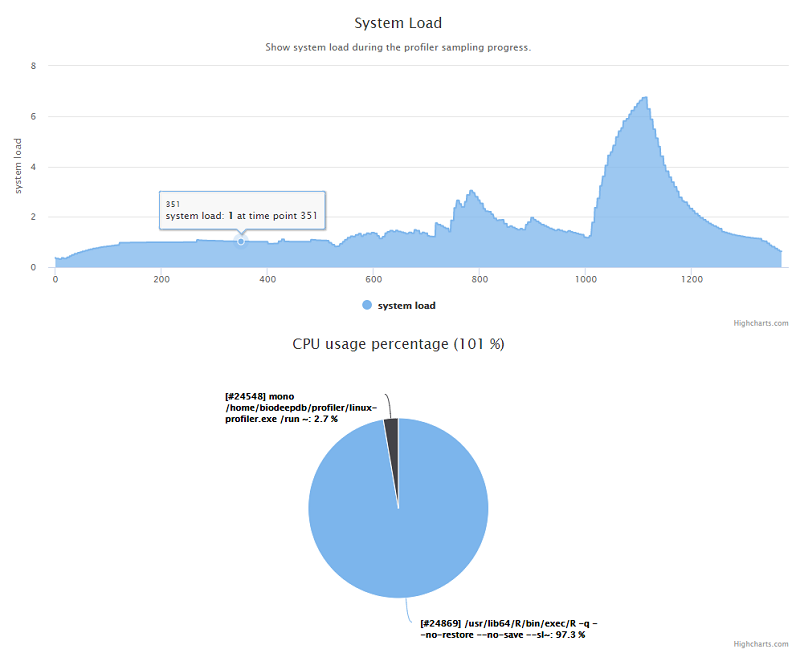
估计阅读时长: 6 分钟https://github.com/xieguigang/linux-profiler 废话不多说,首先给出一个 demo报告链接 给大家看看这个小工具的成品输出。 在去年的工作中,因为公司需要购买新的服务器做集群计算,需要一个工具来记录之前的服务器在数据分析上的性能瓶颈。于是花了两天的时间赶出来了这个专门应用于Linux系统的性能记录工具。这个小工具是一个开源项目,大家可以在Github上阅读这个开源项目(linux-profiler)的源代码。 Order by Date Name Attachments systemLoad • 53 kB • 810 […]
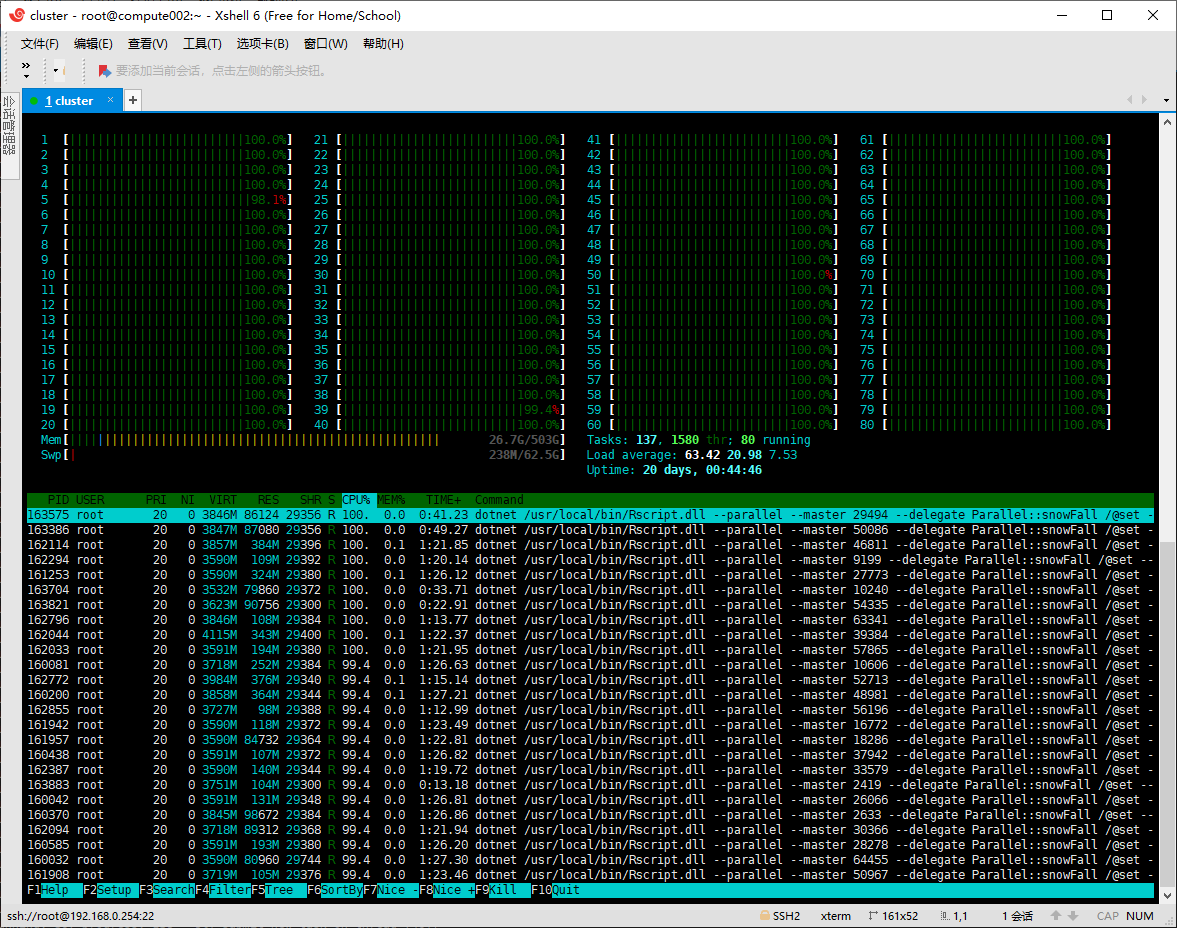
估计阅读时长: 5 分钟https://github.com/xieguigang/Darwinism 最近在做一个代谢组学的数据分析程序,由于需要被分析的质谱原始数据的计算量非常大,所以肯定会需要上并行计算。在并行计算中,分为两种模式:线程并行以及进程并行。 关于如果选择脚本代码的并行模式,我在这里借用了matlab文档网站里面的一张图来给大家做参考: 《Choose Between Thread-Based and Process-Based Environments》 Order by Date Name Attachments super_computing • […]
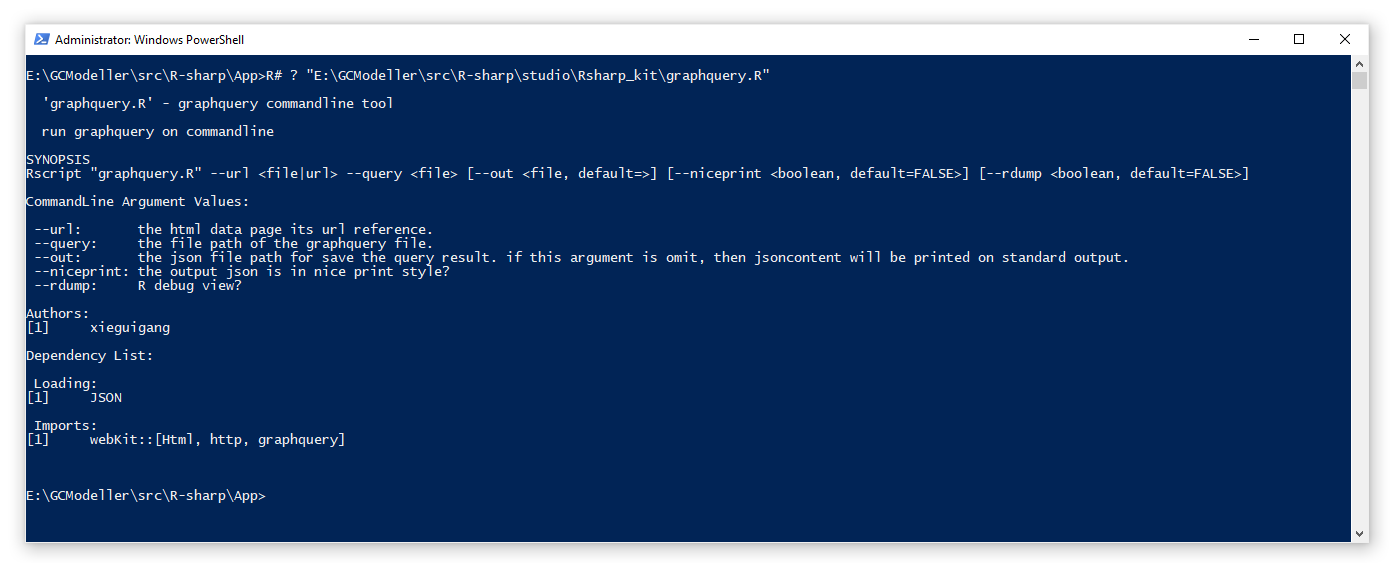
估计阅读时长: 9 分钟前段时间由于工作的需要,会需要从一些网站上抓取数据用来做数据分析。在原来我进行网页爬虫开发的时候,一般会需要专门针对网页格式,使用大量的正则表达式进行内容的解析。由于你也知道,VisualBasic语言所开发的程序为一个编译好的Assembly文件,所以假若所需要爬取的网页格式变化了,我们就需要对代码做修改和重新编译。这个时候就会非常的不方便。 Order by Date Name Attachments ea5d2885-bba5-410f-b02b-0589613412ed • 12 kB • 824 click 2021年5月29日graphquery_Rscript • 36 […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?