
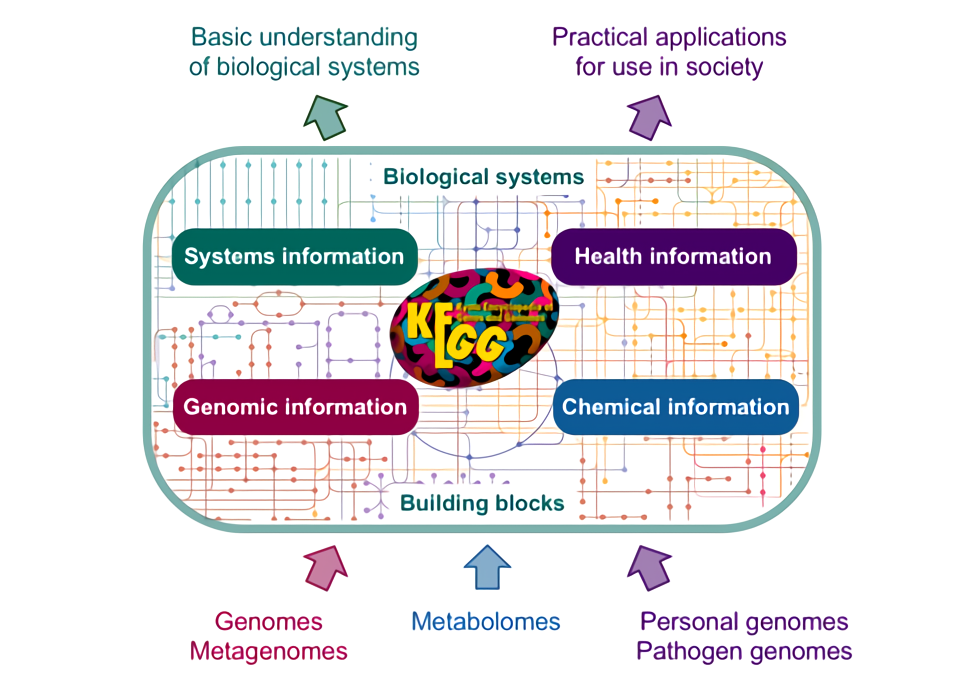
估计阅读时长: 16 分钟KEGG 里面目前并没有“现成的每个 KO 一条代表性序列 FASTA”这种官方序列数据库,假若我们需要基于KEGG数据库中的KO信息的注释,那我们一般会需要自己从 KEGG GENES 里面把每个 KO 对应的基因/蛋白序列抓出来,再按 KO 编号组织成 fasta 集合构建出对应的数据库。基于所建立好的KEGG基因序列数据库,我们就可以实现下面的一些基因注释工作: 在全基因组规模代谢网络重建工作中,进行我们的目标基因组中的代谢网络中的酶节点的直系同源推断,从而将我们的目标基因组中的基因映射到具体的KEGG代谢网络上的节点位置,从而重建出代谢网络模型(使用带有KO编号的蛋白序列做比对注释) 假若我们在进行宏基因组的基因丰度的计算,则可以基于所建立的KEGG基因序列数据库作为参考库,进行宏基因组测序数据中的KO基因丰度的计算(使用带有KO编号的基因序列做比对注释) […]
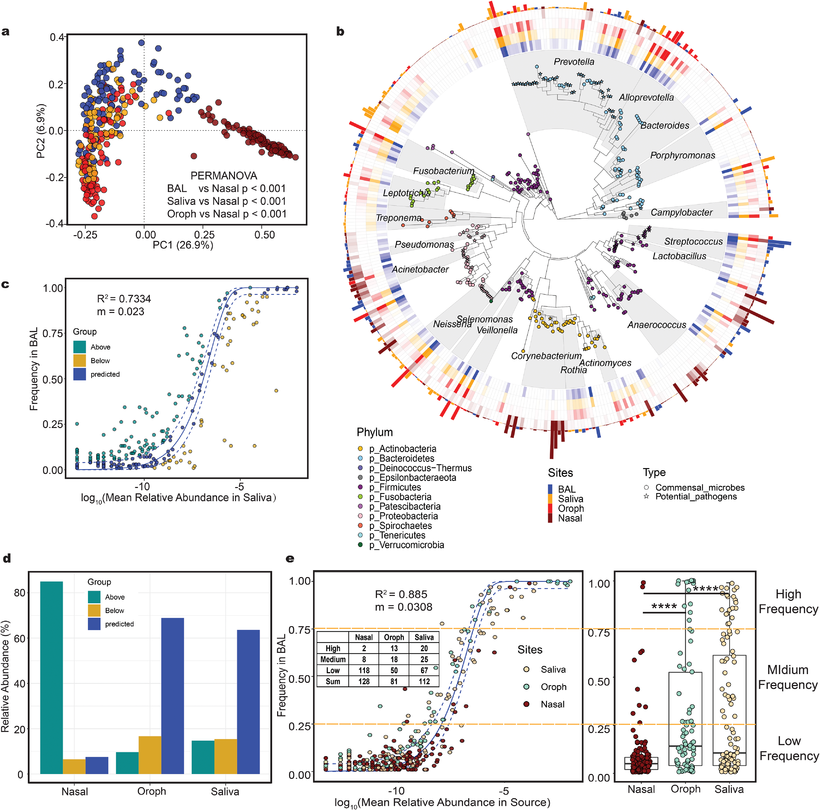
估计阅读时长: < 1 分钟环境中的微生物往往以复杂群落的形式存在,不同物种之间通过代谢相互作用形成协同或竞争关系,共同完成生物地球化学循环、维持生态系统功能。近年来,随着高通量基因组测序技术的发展,研究者可以从环境样本中获取海量微生物基因组数据,为构建基因组尺度代谢模型(Genome-scale metabolic models, GEMs)提供了基础。GEMs将微生物的全基因组注释与生化反应网络相结合,可以用于模拟微生物在特定环境条件下的代谢能力,预测其生长和代谢产物。在单菌株层面,GEMs已被广泛用于解析微生物对环境变化的代谢适应机制、指导代谢工程设计以及预测药物靶点等。在群落层面,通过将多个GEMs耦合,可以研究微生物之间的相互作用,例如通过代谢物交换实现的协同或竞争关系。 Attachments The-taxonomic-composition-of-various-type-samples-and-the-results-of-neutral-model • 500 kB • 238 click 2026年1月4日
估计阅读时长: 6 分钟微生物全基因组代谢网络(Genome-scale metabolic model, GEM)模型的发展历史可追溯至20世纪90年代。1994年,Varma和Palsson在《Applied and Environmental Microbiology》期刊上发表了开创性论文,题为"Stoichiometric flux balance models quantitatively predict growth and metabolic by-product […]
估计阅读时长: 10 分钟https://gcmodeller.org/ 流平衡分析(flux balance analysis)是一种可以用来构建和模拟分析基因组级别的代谢网络的数学方法。流平衡分析是系统生物学(system biology)的一个重要的分析手段。不同于以湿实验的代谢通量分析(metabolic flux analysis, MFA),FBA是用数学方法对代谢网络里的代谢流进行拟合分析。 Order by Date Name Attachments Electron micrographs of […]
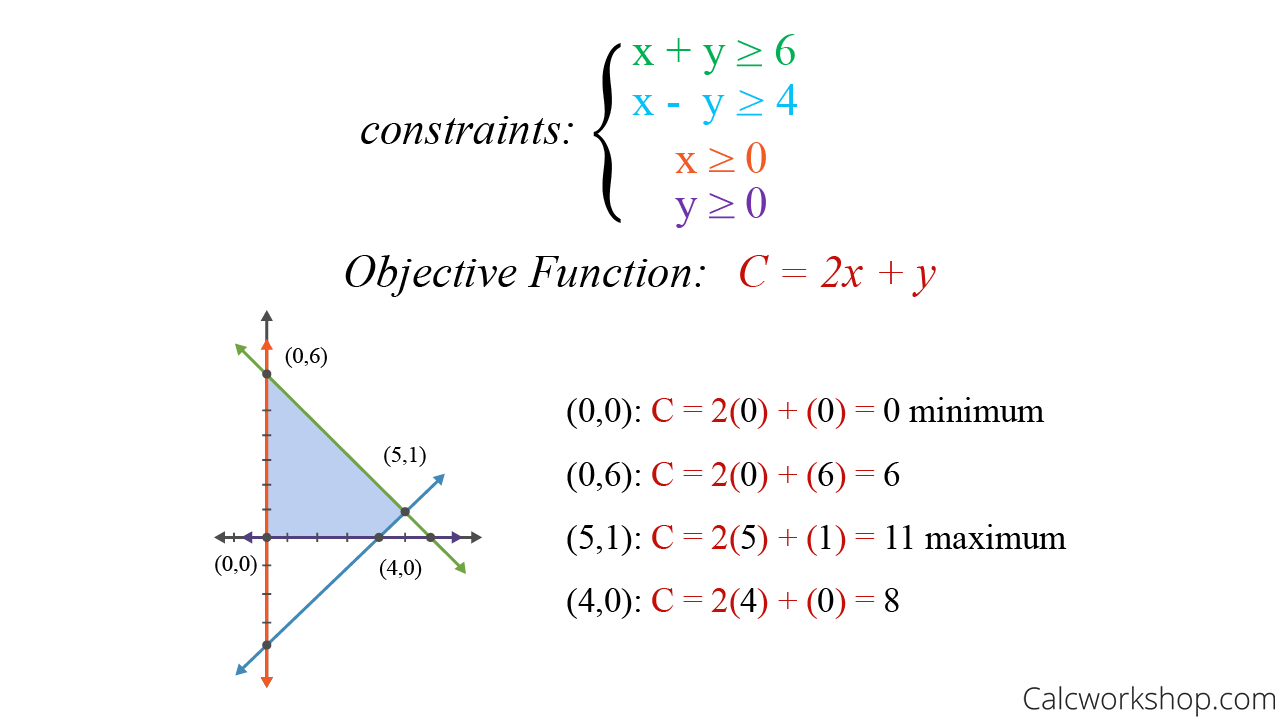
估计阅读时长: 30 分钟https://github.com/xieguigang/sciBASIC/ 线性规划(Linear programming,简称LP)方法起源于20世纪40年代,由美国数学家乔治·丹齐格(George Dantzig)提出,并设计了著名的“单纯形法”。这种优化算法是运筹学中研究较早、发展较快、应用广泛、方法较成熟的一个重要分支,它是辅助人们进行科学管理的一种数学方法。研究线性约束条件下线性目标函数的极值问题的数学理论和方法。通俗点的来讲,就是我们基于这一种数学优化技术,用于在一组线性约束条件下,求解线性目标函数的最大值或最小值(就是在“有限资源”和“一定规则”下,找到“最佳方案”的一种方法)。 Order by Date Name Attachments linear-programming-example • 22 kB • 864 click […]

估计阅读时长: 15 分钟进行生物化学代谢反应网络的模拟计算,可以分为三种技术路线:基于线性规划做优化的FBA方法,基于常微分方程组求解的动力学模拟方法,以及最近发展的基于图神经网络做模拟计算的深度学习计算方法。在下面的表格中,在这里进行比较和总结了上面所提到的三种计算分析方法各自的计算原理和应用领域: 计算方法 原理 优势 适用场景 通量平衡分析(FBA) 基于约束条件(如化学计量矩阵、酶容量限制)和线性规划,在假设代谢网络处于稳态(即代谢物浓度不变)的前提下,计算代谢通量的分布,通常以最大化特定目标(如生物量生长)进行优化 1. 无需详细的酶动力学参数,特别适合大规模网络研究。2. 计算速度快,可系统性地预测基因敲除或环境扰动下的表型变化。3. 广泛应用于指导代谢工程,优化目标产物合成。 追求快速评估和全局优化:如果你的研究目标是在基因组尺度上快速评估微生物在不同条件下的生长或产物合成潜力,并且难以获取详细的动力学参数,FBA是一个非常实用的起点 动力学模拟 基于质量作用定律等构建常微分方程组(ODEs),描述每个代谢物浓度随时间变化的动力学过程,通过数值方法求解方程组 1. 能够捕捉代谢物浓度和通量的瞬态动态变化,揭示更精细的调控机制2. […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?