

估计阅读时长: < 1 分钟https://github.com/rsharp-lang/R-sharp 前言 经过了2021年一年时间的奋斗,目前R#脚本语言环境终于可以算是能够支撑起比较完整的数据分析流程了。在2021年这段时间,我为R#脚本语言环境大概做了以下几件我认为是比较里程碑式的工作: 建立起了一个比较成熟的脚本打包系统 仿照R语言引入了ggplot和ggraph类似的作图系统 借助于mzkit的开发,将R#语言成功的应用于商业化的质谱数据分析产品之中 为了扩大R#语言环境的受众,在2022年初,也就是这个月内,我相继为Python语言和Julia语言添加了对R#语言环境的支持。下面我们就来聊聊在R#语言环境中的对上面所提到的两种语言的支持。 Order by Date Name Attachments programming • 262 kB […]
估计阅读时长: 18 分钟https://github.com/rsharp-lang/R-sharp/tree/master/studio/RData 如果我们需要将上游的R数据分析环境之中的数据集串流至下游的R#数据分析环境之中,构建出一个不同的数据分析环境混合在一块的自动化数据分析流程。我们一般会需要将上游的R环境之中的数据符号对象以RData的格式串流到下游环境中,下游环境进行反序列化加载数据到环境中执行相应的分析。例如在下游执行定制化程度更高的数据作图,将数据以在上游R环境中比较困难实现的其他二进制文件格式进行保存,或者进行分布式的跨物理机的集群化计算,等等用于实现单纯依靠R环境所比较困难实现的功能。 从上一篇博客文章之中我们比较下详细的了解了RData数据文件的文件格式以及对应的读取操作。在这篇文章之中我们来了解如何基于我们通过对RData文件读取操作所获取得到的链表数据进行反序列化操作,将R环境之中的数据集串流加载到下游的R#数据分析环境之中。 Order by Date Name Attachments rstudio-og-fb-1-1024x538 • 39 kB • 992 click 2021年12月4日read-vector […]
估计阅读时长: 19 分钟https://github.com/rsharp-lang/R-sharp/tree/master/studio/RData 在最近的工作中,需要将Docker容器内的R环境之中的数据集无缝的串流到下游的.NET Core数据分析环境之中,基于.NET Core代码库进行数据可视化之类的操作。目前在R环境与.NET Core环境之间进行交互仅存在有一个比较出名的R.NET项目。但是对于使用R.NET项目而言,我们只能够在.NET Core环境之中调用R环境做数据分析,并不能够实现R环境调用.NET Core数据分析环境。并且R.NET项目必须要依赖于R环境对应的库文件,所以使用R.NET并不能够满足我们在Docker容器间进行R数据分析环境与.Net Core数据分析环境之间的无缝衔接。 Order by Date Name Attachments RStudio_Logo • 185 […]

估计阅读时长: 10 分钟https://github.com/xieguigang/ms-imaging Order by Date Name Attachments HR2MSI_mouse_urinary_bladder_S096_RGB • 7 MB • 1089 click 2021年11月13日peerj-cs-07-585 • 16 […]
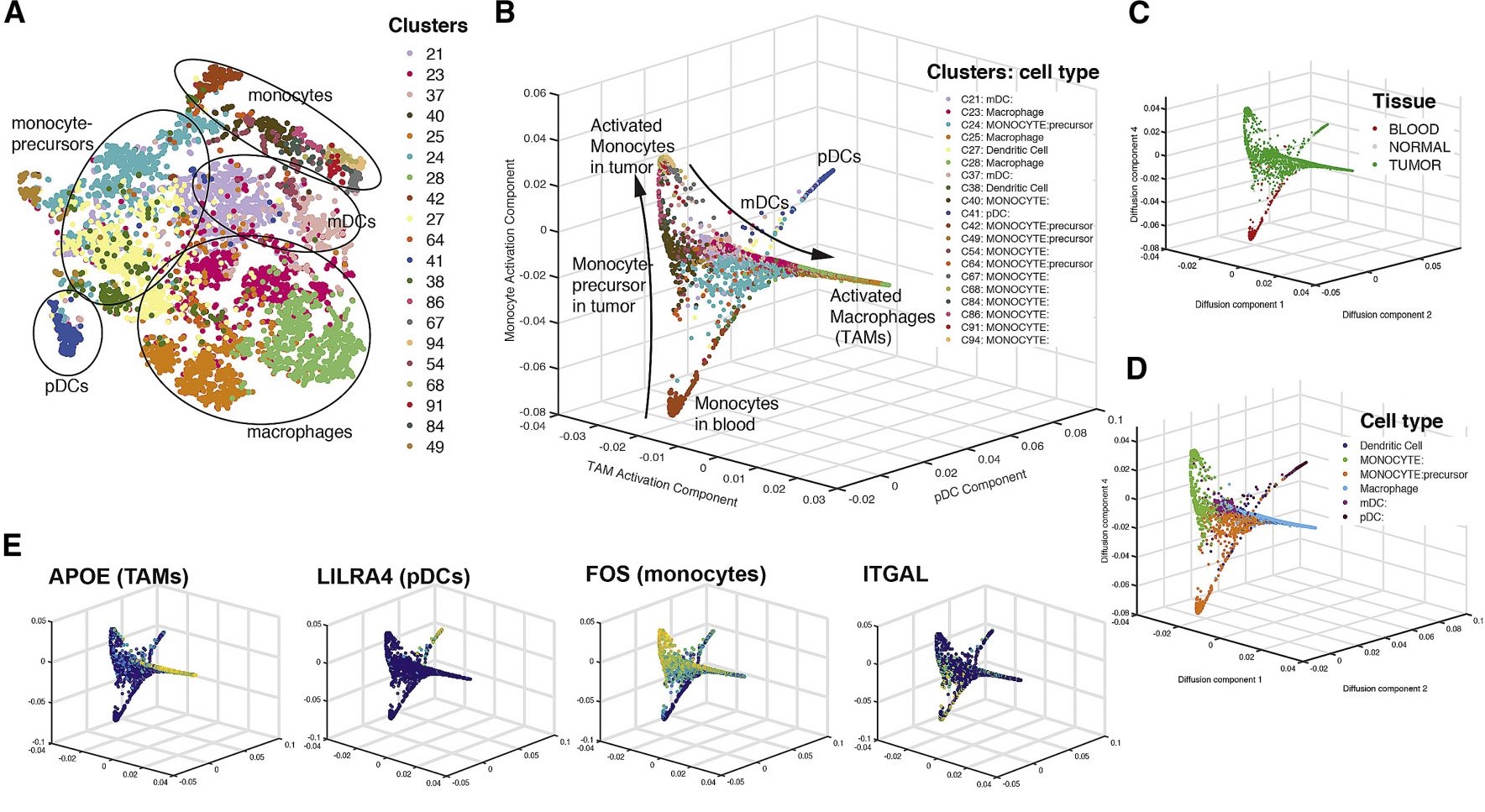
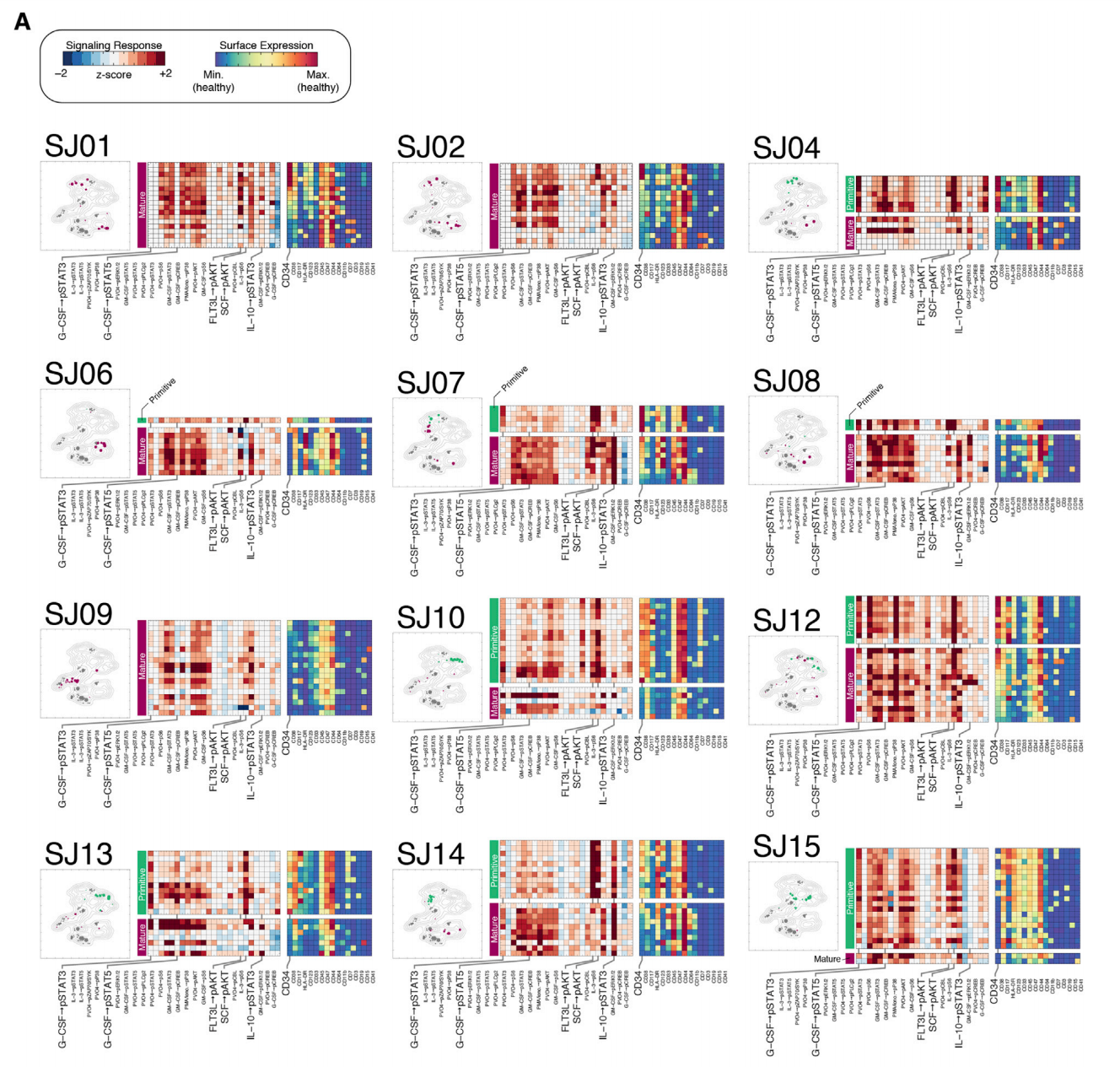
估计阅读时长: 14 分钟https://github.com/rsharp-lang/ggplot 之前在阅读一篇单细胞组学数据分析的文献,觉得在文献之中有一些三维散点图用于展示降维聚类结果的效果非常的好看。于是自己在R#语言之中的ggplot程序包的2D绘图的功能基础之上,进行了三维图形数据可视化功能的开发。 (A) t-SNE map projecting myeloid cells from BC1-8 patients (all tissues). Cells are colored […]

估计阅读时长: 9 分钟https://github.com/rsharp-lang/Rnb 之前使用Python脚本进行编写代码的时候,十分的羡慕Python脚本可以基于ipynb记事本进行文档化的编码。在之前R#脚本是缺少相关的代码库模块将可执行的R#脚本渲染成可视化文档。但是经过几天的开发工作时候,现在R#脚本编程已经具备有了文档化编程的基本框架了。 Order by Date Name Attachments 01510007-school-notebook • 32 kB • 966 click 2021年10月30日renderHtml_cli • […]
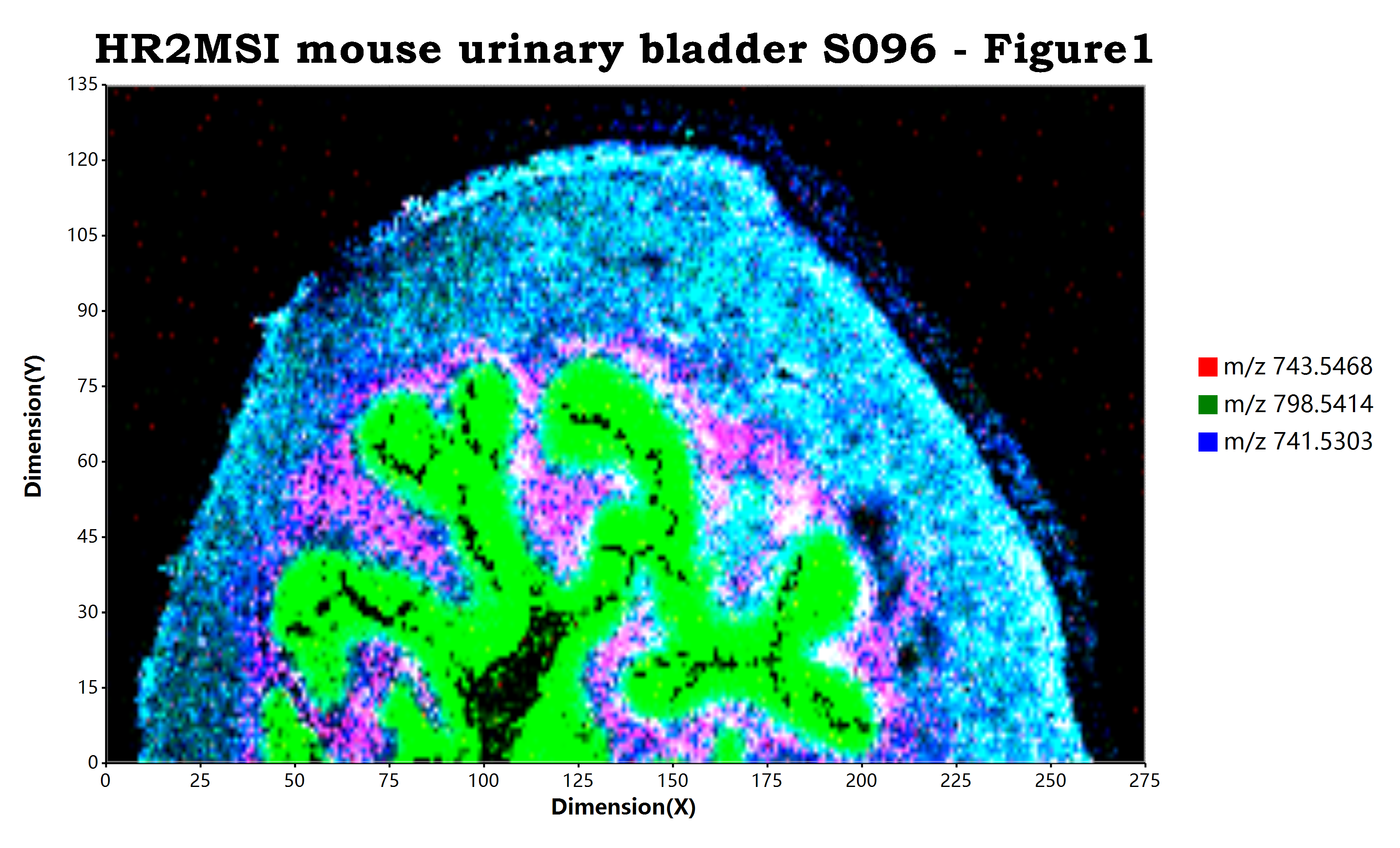
估计阅读时长: 11 分钟https://github.com/xieguigang/ms-imaging Mass spectrometry imaging ( MSI) is a technique used in mass spectrometry to visualize the […]
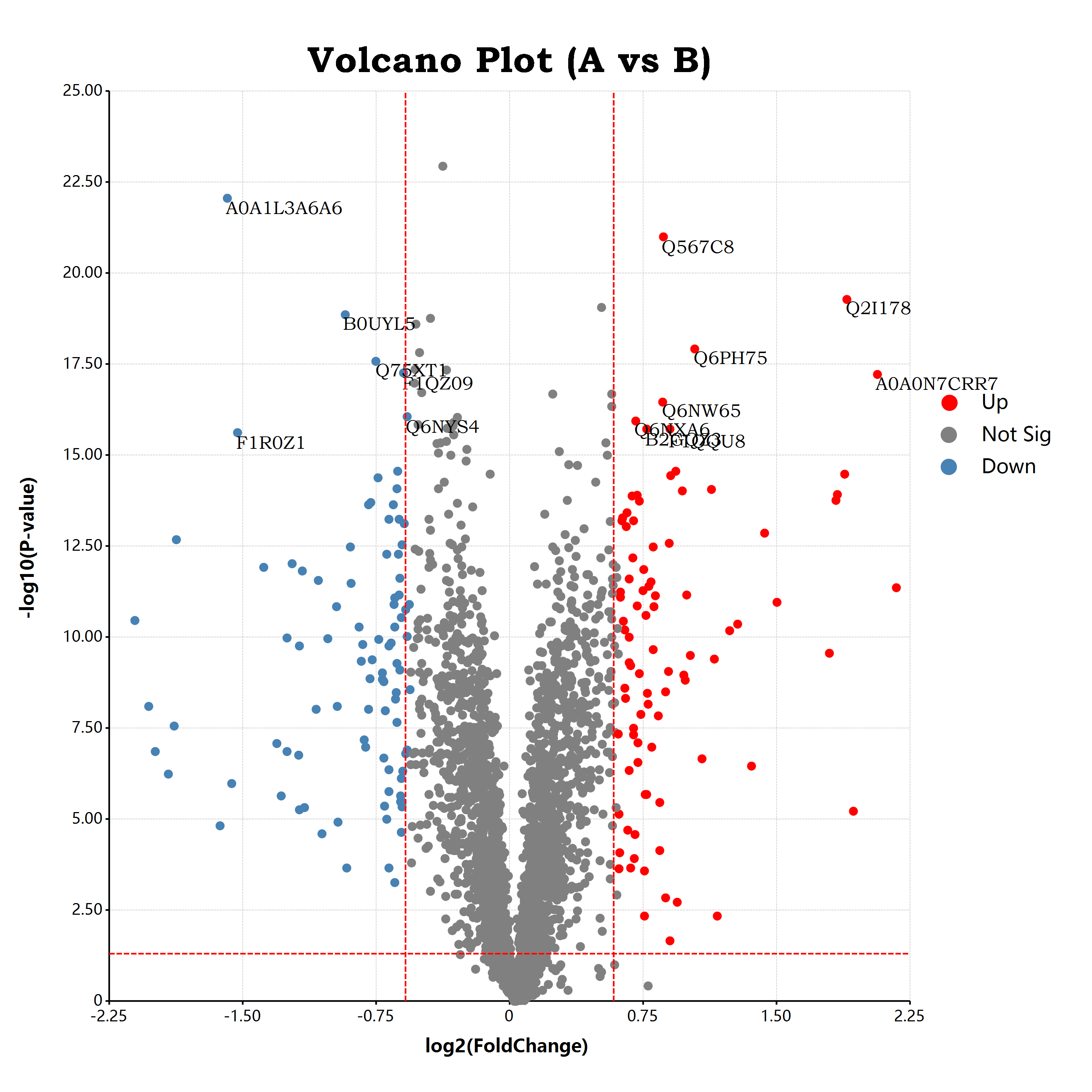
估计阅读时长: 17 分钟https://github.com/rsharp-lang/ggplot 接上一篇博客文章中谈到,我们已经通过R#语言之中的ggplot程序包绘制出了一个可以使用的火山图。在这里,我们将会通过在火山图上添加更多的可视化元素来为大家介绍R#语言之中的ggplot程序包的进阶使用方式。 Order by Date Name Attachments volcano • 651 kB • 1116 click 2021年10月9日volcano • […]
估计阅读时长: 11 分钟https://github.com/rsharp-lang/ggplot 在生物信息学中的组学数据分析领域内,有一个非常常见的数据可视化图表:应用于可视化两两组别比对结果的火山图。在火山图之中,X坐标轴一般是log2FC,纵坐标Y轴,则一般是t检验的pvalue的-log10转换之后的值。由于fold change有大于1的值,A/B大于1,表示A的表达量高于B的表达量,反之小于一表示A的表达量低于B的表达量。这样子fold change经过log2转换之后,就会出现负数,散点一般呈轴对称分布在X=0的位置周围。这样子绘制出来的散点图就有点类似于火山喷发的样子了。 Order by Date Name Attachments a679af1eb9ffbfbad48c18d563ea51f3 • 45 kB • 1020 click […]
估计阅读时长: 7 分钟https://github.com/rsharp-lang/ggplot 一张统计图形就是从数据到几何对象(geometric object, 缩写为geom, 包括点、线、条形等)的图形属性(aesthetic attributes, 缩写为aes, 包括颜色、形状、大小等)的一个映射。此外, 图形中还可能包含数据的统计变换(statistical transformation, 缩写为stats), 最后绘制在某个特定的坐标系(coordinate system, 缩写为coord)中, 而分面(facet, 指将绘图窗口划分为若干个子窗口)则可以用来生成数据中不同子集的图形。 […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?