
文章阅读目录大纲
EC Number是国际酶学委员会(IUBMB)制定的一套酶分类编号体系,EC Number采用层级分类法,由4个数字组成,分别代表酶的大类、亚类、亚亚类和序号。例如,“EC 1.1.1.37”中,第一个“1”表示氧化还原酶大类;第二个“1”表示作用于CH-OH基团;第三个“1”表示以NAD+或NADP+为受体的酶;第四个“37”表示特定酶苹果酸脱氢酶。这种层次结构意味着EC编号蕴含了丰富的功能信息,包括酶催化的反应类型和底物/机制。将EC Number嵌入为向量,有助于我们利用机器学习模型进行功能预测、相似性分析等。
基于之前的一篇文章《TF-IDF与N-gram One-hot文档嵌入算法原理》的学习,我们了解到可以将生物序列通过分解为kmer,组成单词集合用来表示一个文档。从而将长度各异的生物序列嵌入为长读一致的数值向量,进而可以用于后续的各种数据处理工作中。在这里,假设我们将基因组中的所有基因提取出来,然后通过blast比对的方式将基因注释到对应的ec number编号,既可以将某一个基因组使用一个ec number的集合来表示。通过这样子的数据表示方法,我们就可以将任意一个大小各异,基因组成不同的基因组都嵌入为具有相同维度特征的数值向量用于机器学习建模之类的工作。
数据准备工作:基因组蛋白质序列提取
首先,我们可以基于下面的一段R#脚本,从ncbi genbank数据库文件中提取出需要做注释的蛋白序列,以进行后续的blastp搜索注释工作,生成基因组的向量化嵌入所需要的基础数据。
require(GCModeller);
imports "GenBank" from "seqtoolkit";
imports "bioseq.fasta" from "seqtoolkit";
# a local repository directory that contains multiple
# genbank assembly files.
# and scan the genbank assembly files inside this
# repository directory via list.files
let repo = relative_work("./genomes");
let genbank_repo = list.files(repo, pattern = c("*.gbff"));
# read genbank assembly files
let genbank_asm = GenBank::load_genbanks(genbank_repo);
# open fasta sequence file writer stream
let prot = open.fasta(relative_work("./prot.fasta"),
read = FALSE, delimiter = " ");
for(let gb in genbank_asm) {
# scan all genbank assembly data and then extract the
# protein fasta sequence from the genomics annotation data
message(gb |> taxonomy_lineage() |> toString());
write.fasta(seq = protein_seqs(gb, title = "<gb_asm_id>.<locus_tag> <lineage>"), file = prot);
}
# close file stream
close(prot);运行上面的脚本,可以从所下载的ncbi genbank数据库文件中提取出基因组注释得到的蛋白质序列信息。这些蛋白质的fasta序列标题格式如下所示:
在上面的fasta序列提取结果中,为了后续方便构建基因组的EC Number注释模型,在这里我们将GenBank的ID和物种的分类信息提取出来,放入到fasta序列的标题中。按照相同的方法,我们可以从UniProt数据库中,也提取出类似的具有EC Number编号注释的蛋白序列数据库:
blastp搜索注释
很简单的,我们按照下面的blastp命令行,将我们所导出来的需要进行注释的基因组中的蛋白质序列和我们的EC Number的序列数据库进行比对,提取对应的酶注释信息:
/opt/ncbi_blast/bin/blastp \
-query ./prot.fasta \
-db ./ec_numbers.faa \
-evalue 1e-5 \
-out ./protein_ec.txt \
-num_threads 512
大概经过半个小时的比对,我们可以将所分析的一批基因组都完成对应的EC Number的注释搜索工作。blastp的原始结果输出文件比较大,大约是246GB,然后我们就可以通过下面的GCModeller脚本进行比对结果的数据提取工作:
require(GCModeller);
imports "annotation.workflow" from "seqtoolkit";
# commandline input
[@info "the blastp output in text file format."]
[@type "text"]
let rawinput as string = ?"--blast_output" || stop("no blast output raw data file provided!");
let output_table as string = file.path(dirname(rawinput), `${basename(rawinput)}-hits.csv`);
#' Export result hits of blastp output
let exportHits = function(outputText) {
using table as open.stream(output_table, ioRead = FALSE, type = "SBH") {
# parse the raw blastp search result text output
# as the protein hits table
read.blast(outputText, type="prot")
|> blasthit.sbh(keepsRawName = TRUE)
# then save the protein hits table into target file stream
|> stream.flush(stream = table)
;
}
}
# call this data table export function
exportHits(outputText = rawinput);
上面的R#脚本是单线程执行的,每秒钟大约解析10MB的原始数据,blastp数据全部处理完大概会花费掉7个半小时。处理完的原始注释结果表格同样也比较大,大约为29GB。
所提取出来的结果表格,可以直接通过R#解释器程序进行预览:
R# --head --file ./protein_ec-hits.csv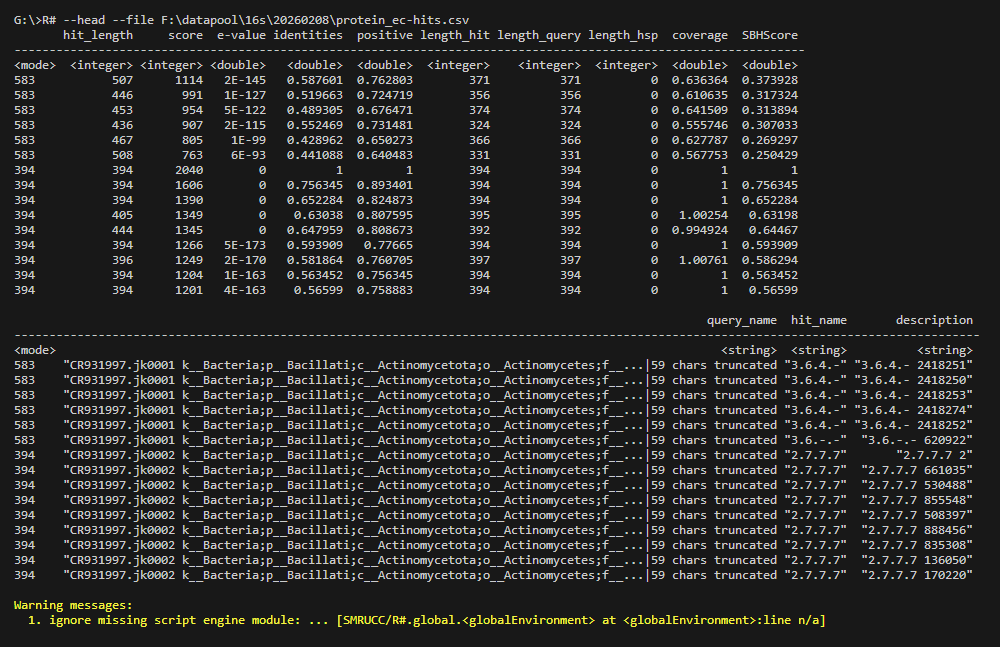
搜索结果解析为EC Number
在上面所提取出来的原始blastp搜索结果表格的基础之上,可以进一步的通过下面的R#脚本,将原始的搜索结果处理为对应的EC Number的映射关系结果,完成基因组中的基因对EC Number的注释工作:
require(GCModeller);
imports ["annotation.workflow", "annotation.terms"] from "seqtoolkit";
# read the raw blastp hits table and then
# make ec number term assignment
relative_work("protein_ec-hits.csv")
|> read.besthits()
|> assign_terms()
|> write.csv(relative_work("ec_terms.csv"))
;运行完上面的脚本后,既可以将原始的blastp搜索结果表格解析为下面的EC Number对基因组中的基因的映射结果:

require(GCModeller);
require(jsonlite);
imports "annotation.terms" from "seqtoolkit";
# make genomics annotation data view
let metabolic = relative_work("ec_terms.csv")
|> read_rankterms()
|> make_vectors()
;
# dump the genome metabolic annotation result
# into json file
writeLines(jsonlite::toJSON(metabolic),
con = relative_work("metabolic_enzymes.json"));将上面的EC Number的注释结果按照基因组合并之后,就可以得到了下面的基因组代谢酶的注释结果:

在上面得到的基因组代谢酶注释结果中,可以看到我们的注释结果的数据结构由基因组的ID信息和注释得到的ec number集合所构成。在这里我们可以将得到的基因组看作为ec number单词集合的文档数据,那么基于此数据结构,我们就可以应用之前的文章中所介绍的TF-IDF向量嵌入算法将不同大小不同基因组成成分的基因组嵌入为相同维度的数值向量结果。从而用于后续的机器学习建模工作。
基因组注释结果进行TF-IDF向量化嵌入
基于所构建的TF-IDF算法核心,可以封装出如下的基因组代谢酶注释结果的向量化嵌入算法模块:
Imports Microsoft.VisualBasic.Data.Framework
Imports Microsoft.VisualBasic.Data.NLP
Imports SMRUCC.genomics.Interops.NCBI.Extensions.Pipeline
Public Class GenomeVector : Implements INamedValue
Public Property assembly_id As String Implements INamedValue.Key
Public Property taxonomy As String
' metabolic annotation term collection
Public Property terms As Dictionary(Of String, Integer)
Public ReadOnly Property size As Integer
Get
If terms Is Nothing Then
Return 0
Else
Return terms.Values.Sum
End If
End Get
End Property
Public Overrides Function ToString() As String
Return taxonomy
End Function
End Class
Public Class GenomeMetabolicEmbedding
ReadOnly vec As New TFIDF
ReadOnly taxonomy As New Dictionary(Of String, String)
Public Sub Add(genome As GenomeVector)
Call vec.Add(genome.assembly_id, genome.terms)
Call taxonomy.Add(genome.assembly_id, genome.taxonomy)
End Sub
Public Function AddGenomes(seqs As IEnumerable(Of GenomeVector)) As GenomeMetabolicEmbedding
For Each annotation As GenomeVector In seqs
Call Add(annotation)
Next
Return Me
End Function
Public Function TfidfVectorizer(Optional normalize As Boolean = False) As DataFrame
Call $"Make metabolic embedding with: ".info
Call $" * {vec.N} genomes".debug
Call $" * {vec.Words.Length} total enzyme terms".debug
Call VBDebugger.EchoLine("")
Dim df As DataFrame = vec.TfidfVectorizer(normalize)
Call df.add("taxonomy", From id As String In df.rownames Select taxonomy(id))
Return df
End Function
''' <summary>
''' n-gram One-hot(Bag-of-n-grams)
''' </summary>
''' <returns></returns>
Public Function OneHotVectorizer() As DataFrame
Return vec.OneHotVectorizer
End Function
End Class基于上面所封装的TF-IDF算法模块,可以实现出下面的API函数用于R#脚本编程,实现针对基因组的向量化嵌入操作:
''' <summary>
''' make embedding of the genomics metabolic model
''' </summary>
''' <param name="annotations"></param>
''' <param name="L2_norm"></param>
''' <param name="env"></param>
''' <returns></returns>
<ExportAPI("tfidf_vectorizer")>
Public Function tfidf_vectorizer(<RRawVectorArgument>
annotations As Object,
Optional L2_norm As Boolean = False,
Optional union_contigs As Integer = 1000,
Optional env As Environment = Nothing) As Object
Dim genomes As pipeline = pipeline.TryCreatePipeline(Of GenomeVector)(annotations, env)
If genomes.isError Then
Return genomes.getError
End If
Dim genome_asm As IEnumerable(Of GenomeVector) = GenomeVector.GroupByTaxonomy(
genomes.populates(Of GenomeVector)(env), union_contigs)
Dim idf As GenomeMetabolicEmbedding = New GenomeMetabolicEmbedding().AddGenomes(genome_asm)
Dim vec = idf.TfidfVectorizer(normalize:=L2_norm)
Dim df As New dataframe With {
.rownames = vec.rownames,
.columns = New Dictionary(Of String, Array)
}
Call df.add("taxonomy", vec("taxonomy").vector)
For Each term As String In vec.featureNames
If term <> "taxonomy" Then
Call df.add(term, vec(term).vector)
End If
Next
Return df
End Function基于这里导出来的TF-IDF向量化嵌入工具函数,可以编写出如下R#脚本,实现基于基因组的代谢酶注释结果,完成基因组的向量化嵌入操作:
require(GCModeller);
imports "annotation.terms" from "seqtoolkit";
relative_work("ec_terms.csv")
|> read_rankterms()
|> make_vectors()
|> tfidf_vectorizer()
|> write.csv(relative_work("metabolic_embedding.csv"))
;最终,得到了如下所示的嵌入后的结果矩阵:

向量化嵌入结果可视化
在这里,我们基于UMAP降维的方法对上面所生成的基因组嵌入矩阵进行低维度嵌入操作,将几千个维度的基因组向量嵌入为仅包含有两个维度的向量,基于此低维度嵌入结果,我们可以进行散点图可视化。下面的R#脚本为UMAP低维度嵌入操作的脚本:
require(GCModeller);
require(umap);
imports "taxonomy_kit" from "metagenomics_kit";
let data = read.csv("metabolic_embedding.csv",
row.names = "taxonomy",
check.names = FALSE)
|> select(-1)
;
let result = umap(data, neighbors = 128,
local_connectivity = 2,
method = "Cosine");
let manifold = as.data.frame(result$umap, labels = result$labels);
let taxon = biom_string.parse(rownames(manifold));
manifold[,"phylum"] <- taxonomy_name(taxon, rank = "phylum");
manifold[,"class"] <- taxonomy_name(taxon, rank = "class");
manifold[,"order"] <- taxonomy_name(taxon, rank = "order");
write.csv(result, file = "umap.csv");
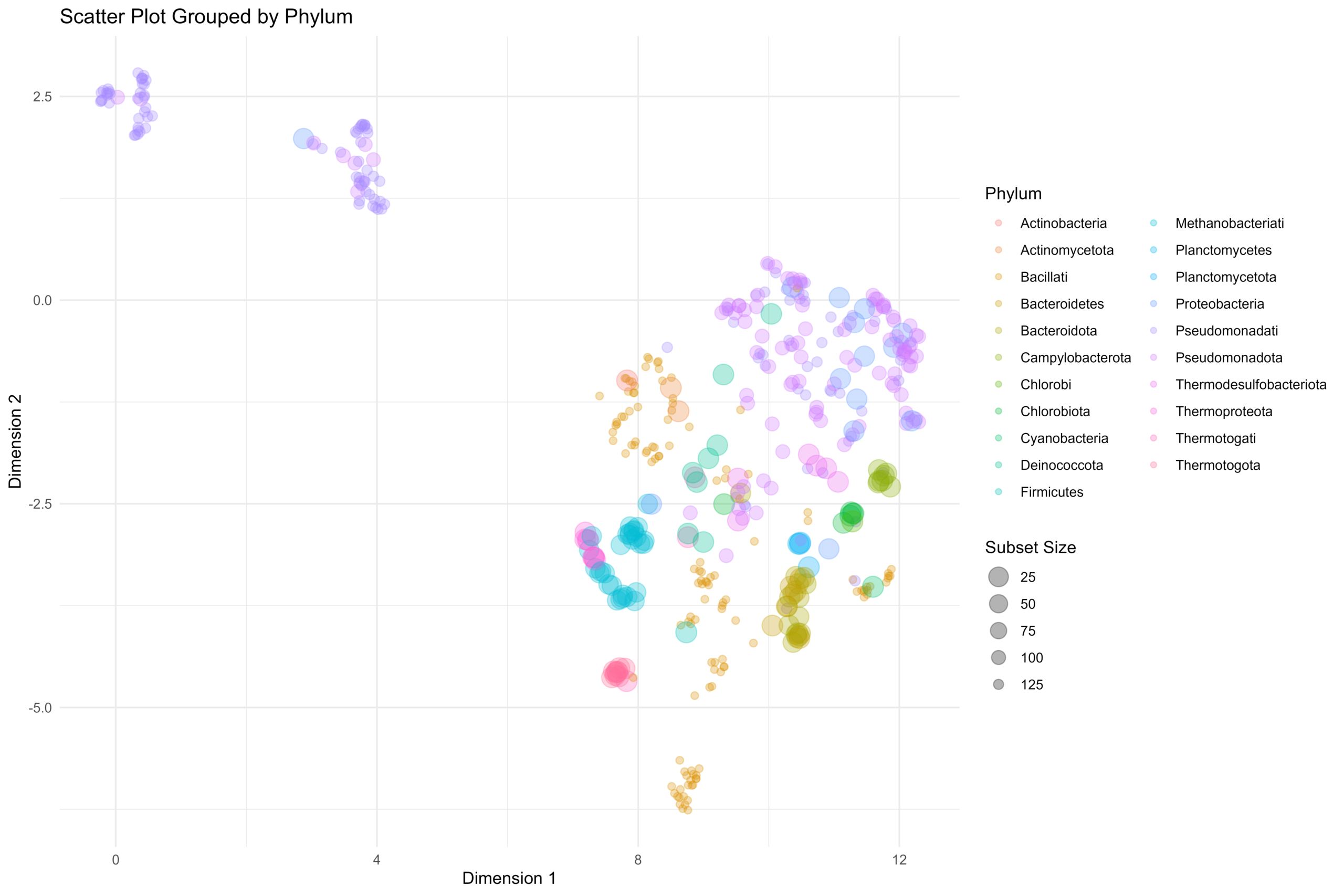
基于所生成的UMAP低维度嵌入结果,可以通过下面的ggplot脚本进行散点图可视化操作。在这里,我们在物种分类的Phylum层级进行散点图的数据分组,不同的Phylum分类的基因组具有各自对应的颜色。散点图中的散点的半径大小与Phylum组中的基因组数量逆相关。
let genome_scatter_viz = function(data) {
# 数据预处理:计算每个 Phylum 分组的大小
# 我们需要给每一行数据都标记上它所属的 Phylum 组有多少个样本
let data_processed = data
|> group_by(phylum)
# 计算每组的数量,并存为新列 subset_size
|> mutate(subset_size = n())
|> ungroup()
;
# 绘制散点图
let p <- ggplot(data_processed, aes(x = dimension_1, y = dimension_2))
+ geom_point(
aes(
color = phylum, # 颜色映射到 phylum
size = subset_size # 大小映射到分组子集大小
),
alpha = 0.3 # 设置全局半透明度为 0.3
)
# 设置大小的映射变换:集合越大,点越小
# trans = "reverse" 会反转坐标轴,即数据值越大(集合越大),图形尺寸越小
+ scale_size_continuous(
name = "Subset Size", # 图例标题
trans = "reverse", # 关键:反转大小逻辑
range = c(2, 6) # 设置点的大小范围(最小点,最大点)
)
# 添加图表标签和主题
+ labs(
title = "Scatter Plot Grouped by Phylum",
x = "Dimension 1",
y = "Dimension 2",
# 颜色图例标题
color = "Phylum"
)
+ theme_minimal()
;
ggsave("scatter_plot.png", plot = p,
width = 12, height = 8,
dpi = 600);
ggsave("scatter_plot.svg", plot = p,
width = 12, height = 8,
dpi = 600);
}
native_r(genome_scatter_viz,
args = list(data = read.csv("umap.csv", row.names = 1, check.names = TRUE)),
require = c("ggplot2", "dplyr")
);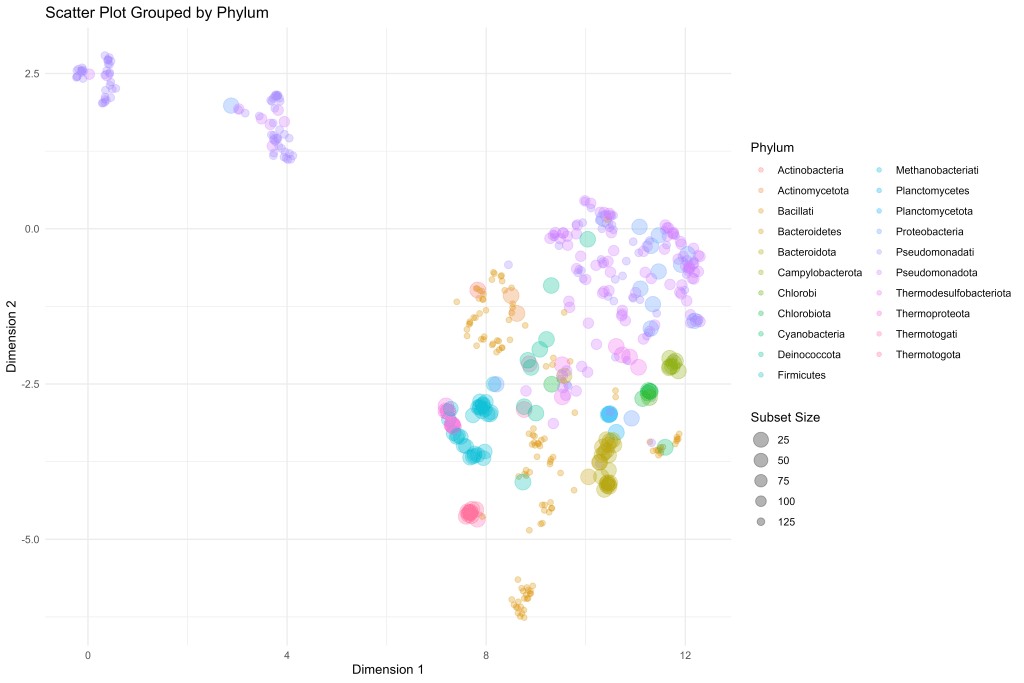
可以看得到,我们得到的基因组的嵌入结果可以很好的对基因组进行物种分类工作。在我们所得到的基因组向量化嵌入结果的散点图上,相同颜色的散点(相同Phylum分类下的基因组)都是相互聚集在一起,说明我们的基因组代谢功能注释向量化嵌入结果可以很好的提取出基因组的代谢特征。
- 单细胞视角下的微生物基因组代谢酶嵌入分析 - 2026年2月25日
- 基因组代谢酶层级嵌入 - 2026年2月23日
- 基因组功能吉布斯LDA主题建模 - 2026年2月23日

5 Responses
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
[…] 最近的工作中我需要按照之前的这篇博客文章《基因组功能注释(EC Number)的向量化嵌入》中所描述的流程,将好几十万个微生物基因组的功能蛋白进行酶编号的比对注释,然后基于注释结果进行向量化嵌入然后进行数据可视化。通过R#脚本对这些微生物基因组的蛋白fasta序列的提取操作,最终得到了一个大约是58GB的蛋白序列。然后将这个比较大型的蛋白序列比对到自己所收集到的ec number注释的蛋白序列参考数据库之上。 […]
A very inspiring pipeline for turning EC-based annotations into genome-scale embeddings. Great post! I really enjoyed the clear, end‑to‑end pipeline you presented for converting EC number annotations into fixed‑dimensional genome vectors using TF–IDF and UMAP. there are a few things I particularly liked:
1. Intuitive framing of genomes as “documents”: Treating each genome as a bag of EC terms (i.e., “words” describing metabolic potential) is a clever way to apply classic NLP-style embeddings to comparative genomics. It naturally solves the problem of variable genome size and gene content while preserving functional information.
2. Practical, reproducible workflow from raw genomes to embeddings: The post doesn’t stop at the idea—you walk through the whole process: extracting protein sequences from GenBank, performing large‑scale BLASTp against an EC number sequence database, parsing results, building term tables, and finally training a TF–IDF vectorizer. The R# scripts and GCModeller modules make it straightforward for readers to adapt the approach to their own datasets.
3. Thoughtful use of dimensionality reduction for interpretation: Visualizing the TF–IDF embeddings with UMAP and coloring by taxonomic phylum is a strong proof‑of‑concept. The fact that genomes from the same phylum cluster together suggests that the EC‑based vectors really capture biologically meaningful metabolic signals, not just technical noise.
4. Clean software design and API layer: The GenomeMetabolicEmbedding class and the tfidf_vectorizer API function provide a clean abstraction over the underlying TF–IDF machinery. This makes it easy to plug the method into downstream machine learning pipelines (e.g., classification, regression, or distance‑based analyses).
and some possible extensions that came to mind while reading:
Incorporating hierarchical information of EC numbers (the four‑level enzyme hierarchy) into the term weighting scheme, for example by over‑weighting more informative higher‑level categories or using a hierarchical TF–IDF variant.
Comparing EC‑based embeddings with KO‑ or pathway‑based embeddings to see which representation gives better separation of known phenotypes or taxonomic groups.
Adding a small case study (e.g., phenotypic prediction or outlier detection) to demonstrate the practical benefits of these embeddings in a concrete biological task.
Overall, this is a very well‑written and practical introduction to “functional document embedding” for genomes. It will be a useful reference for anyone working on comparative genomics, metabolic modeling, or machine learning on functional annotation data. Thanks for sharing the code and detailed explanations!
Thank you so much for your thoughtful and encouraging comment! I truly appreciate the time you took to read through the post and provide such detailed, constructive feedback. 🤠
I’m especially glad that the “genome-as-document” analogy resonated with you—it’s a perspective that bridges classical NLP techniques with functional genomics in a surprisingly natural way. Your point about leveraging the hierarchical structure of EC numbers is excellent; in fact, we’re already exploring a hierarchical TF–IDF variant that assigns different weights to each level (e.g., EC 1 vs. EC 1.1.1.37) to better reflect functional specificity. That could indeed enrich the embedding space.
Your suggestions regarding comparative evaluations (e.g., EC vs. KO vs. pathway-based embeddings) and concrete downstream applications like phenotypic prediction are spot-on. We’re currently working on a small case study involving microbial growth phenotype prediction using these embeddings, and early results are promising. I’ll be sure to share updates if it leads to a follow-up post or tool release.
Thanks again for your kind words and insightful ideas—they not only validate the approach but also inspire further improvements. If you ever try this pipeline on your own data or have more thoughts to share, I’d be thrilled to hear about it!
Best regards,