
估计阅读时长: 22 分钟MSA(多序列比对)在生物信息学中的核心目标是:通过把多条同源序列“对齐”,来突出它们之间的相似与差异,从而帮助我们:识别保守区/功能位点、推断进化关系(系统发生)、预测或解释蛋白质/核酸结构、发现共进化与功能模块,以及为后续分析(如模体搜索、结构建模、从头设计等)提供基础。基于多序列比对分析,我们可以通过这种算法,把一堆表面看上去“乱糟糟”的序列,整理成一个可以“逐位点比较”的框架。基于我们所得到的这个框架基础,我们可以进行下游的后续分析,例如: 识别哪些部分是“不能动”的(功能/结构核心); 推断它们是如何“进化而来”的(系统发生); 推测它们在空间中“长什么样”(结构预测与建模); 找出哪些部分“一起变化”(共进化与功能耦合); 并把这些信息封装成模型(HMM、profile)用于大规模搜索与注释。 Order by Date Name Attachments MSA • 174 kB • […]
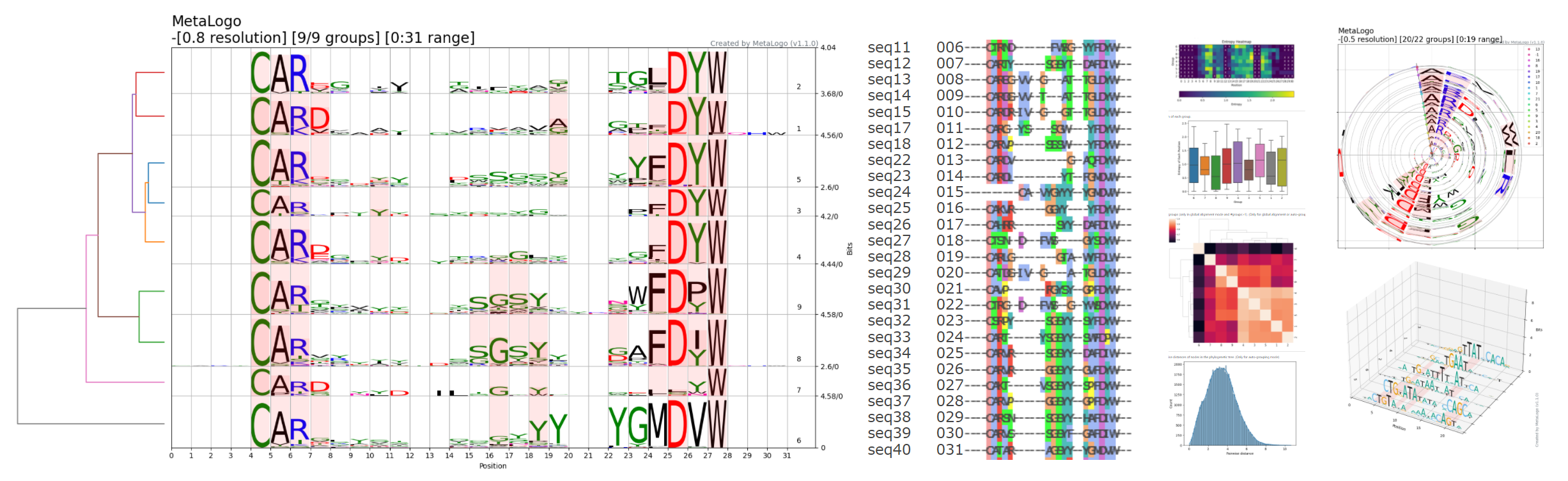
估计阅读时长: 23 分钟Sequence Logo 是一种可视化 DNA 或蛋白质序列保守性的图形表示方法。每个位置(列)上的字母堆叠高度代表该位点的信息含量(以 bits 为单位),而每个字母的高度则与其在该位点出现的频率成正比。高信息量的位置字母堆得高,低信息量的位置则矮甚至接近零。Sequence Logo的绘制遵循信息熵原理,我们可以很直观的通过某一个位置的总高低来了解该处位置的信息含量有多少,高信息量的位置,字母堆的高,一般会出现某一个字符特别高,表明该处非常保守。 位置权重矩阵(Position Weight Matrix, PWM)是描述基因组调控因子结合位点序列模式的核心模型。它通过统计在结合位点序列中每个位置上各核苷酸(或氨基酸)出现的频率,来量化该位置对不同碱基的偏好程度。PWM通常以矩阵形式表示,行对应核苷酸(A、C、G、T/U),列对应序列中的位置,矩阵元素即为该位置该核苷酸相对于背景的权重得分。这一模型简洁且易于计算,因此在转录因子结合位点(TFBS)等调控元件的识别和表征中被广泛采用。 Order by Date Name […]

估计阅读时长: 7 分钟一般而言,进行全基因组的转录表达调控网络的建立,我们需要基于两个数据结果来完成: 目标基因的转录调控位点信息(Motif搜索结果,构成网络之中的节点) 转录调控位点相应的转录调控因子(Motif位点相关的转录调控因子,构成网络之中的边连接) Order by Date Name Attachments Xor • 271 kB • 967 click 2022年6月11日An […]
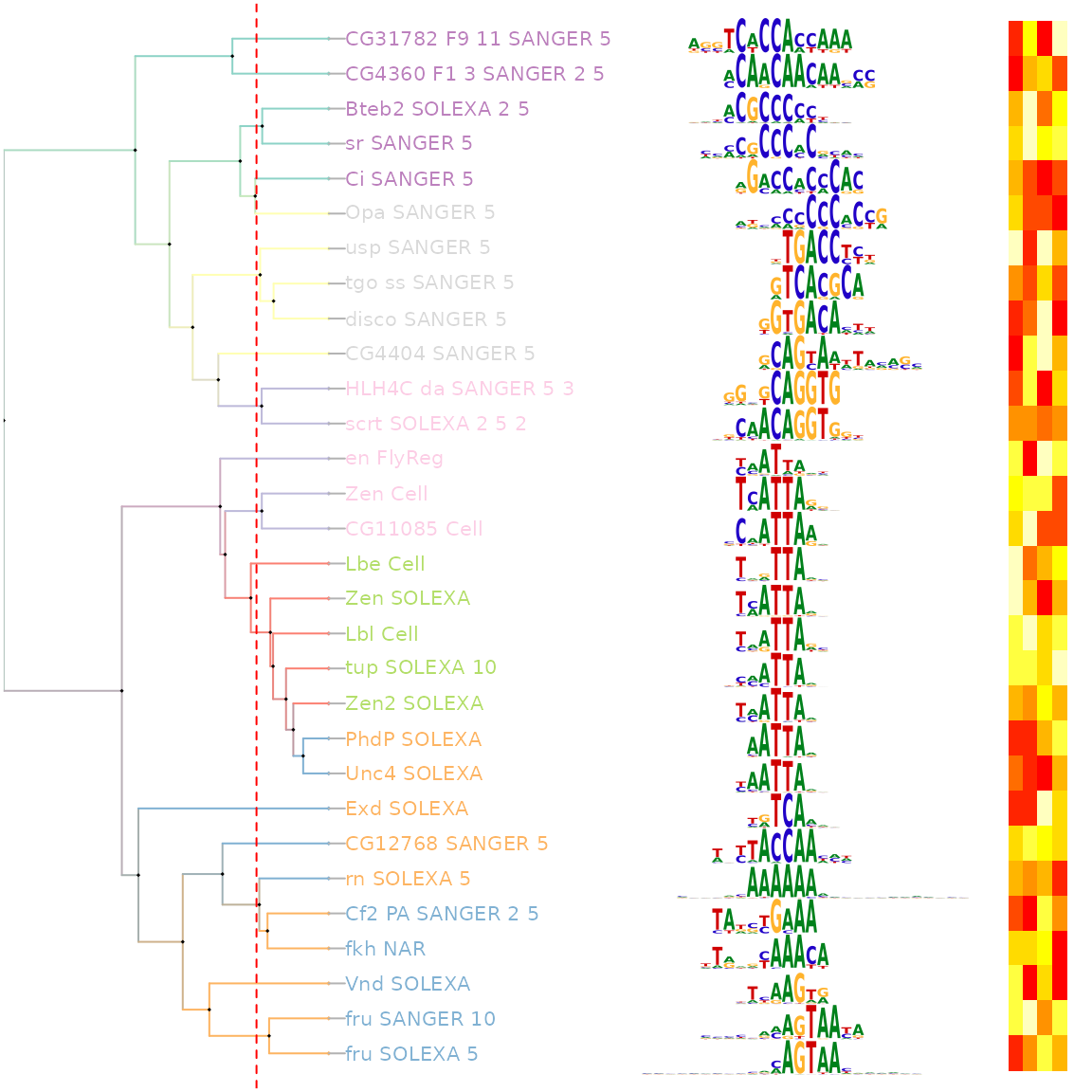
估计阅读时长: 12 分钟Motif是一段典型的序列或者一个结构。一般情况下是指构成任何一种特征序列的基本结构。通俗来讲,即是有特征的短序列,一般认为它是拥有生物学功能的保守序列,可能包含特异性的结合位点,或者是涉及某一个特定生物学过程的有共性的序列区段。比如蛋白质的序列特异性结合位点,如核酸酶和转录因子。 Order by Date Name Attachments Smith-Waterman-Algorithm-Example-Step3 • 8 kB • 972 click 2022年6月7日motifPilesHeatmap-1 • 227 […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?