
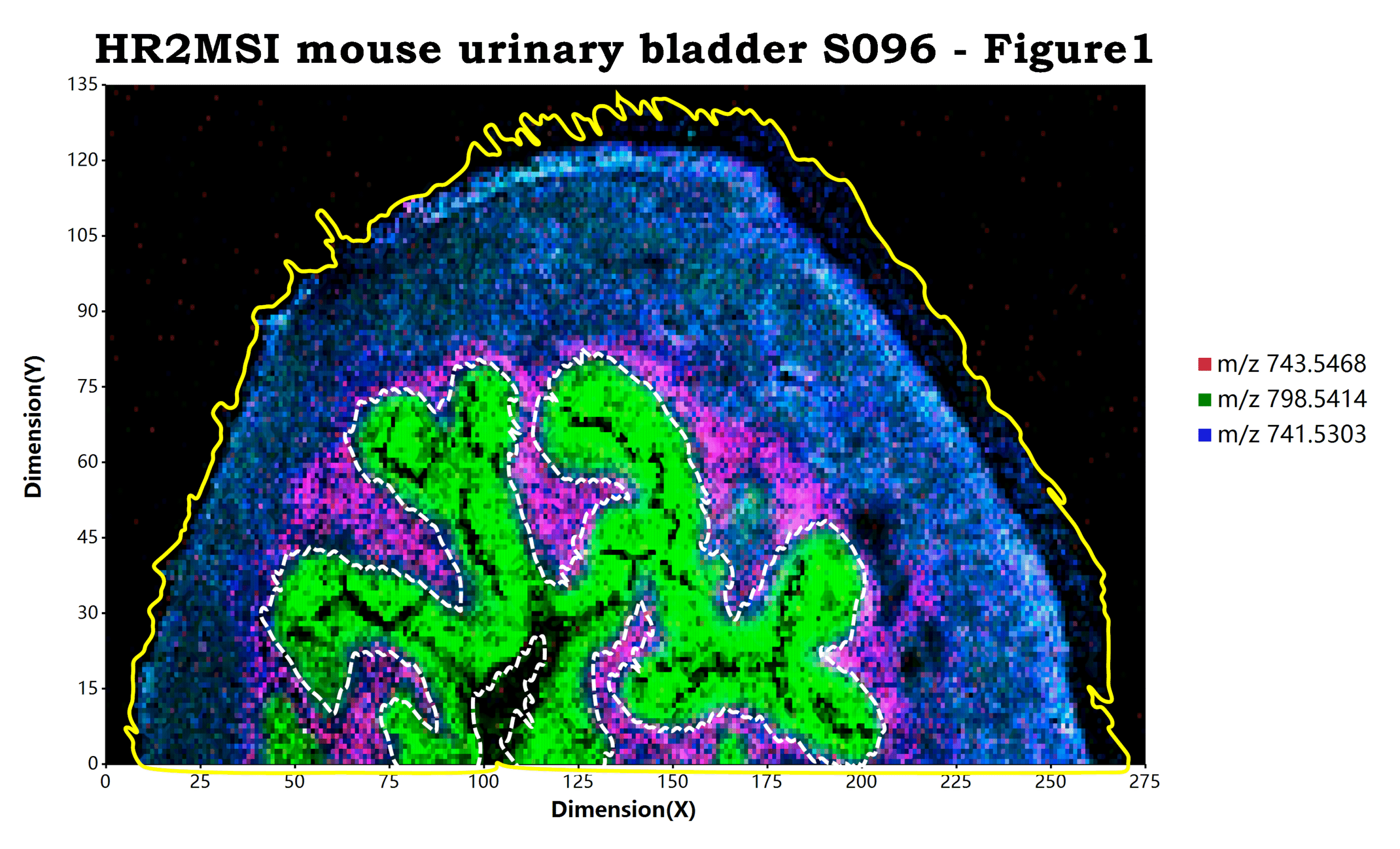
估计阅读时长: 10 分钟目前经过改进和优化之后的基于mzkit代码库底层的msimaging质谱成像软件包在样本可视化上进行了非常多的改进,诸如: 添加样本原始背景叠加 目前进行质谱成像可视化,程序包不仅仅可以使用任意rgb纯色来作为可视化的背景。目前还可以支持直接使用原始数据的背景作为质谱成像的显示背景。进行这个显示的秘诀就在于简单的在脚本中添加一个TIC背景图层:geom_MSIbackground("TIC") ggplot(msi_data, padding = "padding: 200px 600px 200px 250px;") + geom_MSIbackground("TIC") # rendering of […]
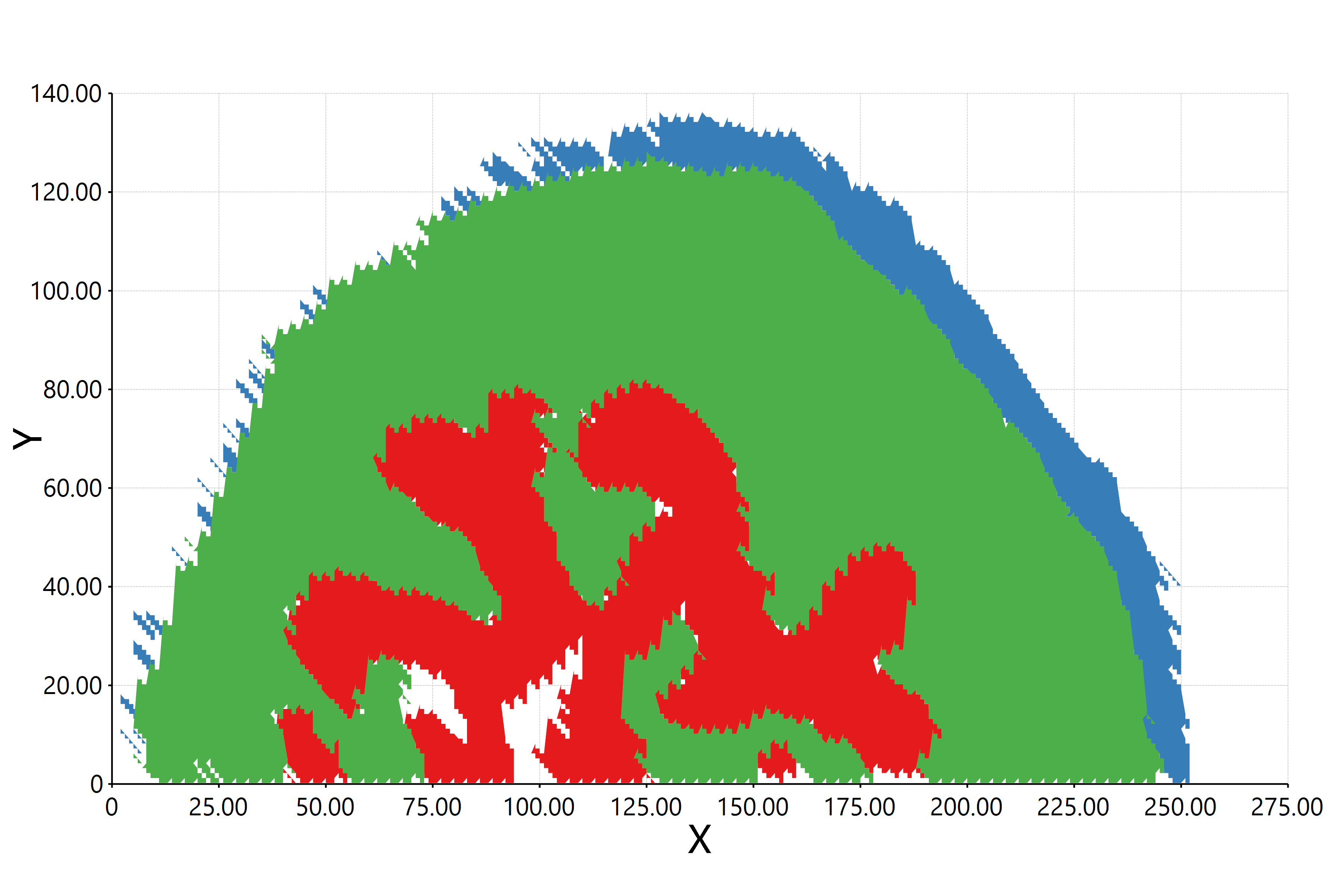
估计阅读时长: 11 分钟https://github.com/xieguigang/sciBASIC 最近在研究实现空间代谢组学中的一些特征区域的自动化划分分割。在得到了特征点集合之后,我们需要根据一些图像处理算法进行特征区域的提取操作。之前,我们尝试过基于绘制等高线图Marching Squares算法的方式来将特征点集合自动转换为特征区域的多边形,实现轮廓扫描获取的功能。但是实现的效果嘛,和实际的区域存在着一些较大的差异。 Order by Date Name Attachments HR2MSI mouse urinary bladder S096 - spatial regions […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?