
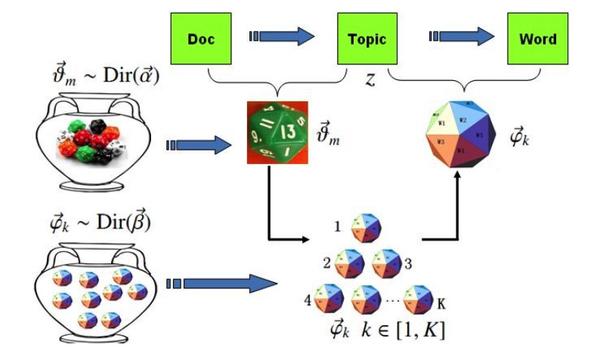
估计阅读时长: 20 分钟LDA(Latent Dirichlet Allocation,潜在狄利克雷分配)是一种用于发现文档集合中潜在主题的生成式概率模型。它假设文档是由多个主题混合而成的,而每个主题又是通过一定的概率分布选择词语生成的。LDA模型包含词、主题和文档三层结构,通过概率生成过程模拟文档的形成。Gibbs LDA 的核心在于使用吉布斯采样方法来推断这些隐藏的主题分布。 Attachments v2-883ac9db7f1cbd7325b2450cd225a897_b • 29 kB • 379 click 2026年2月23日

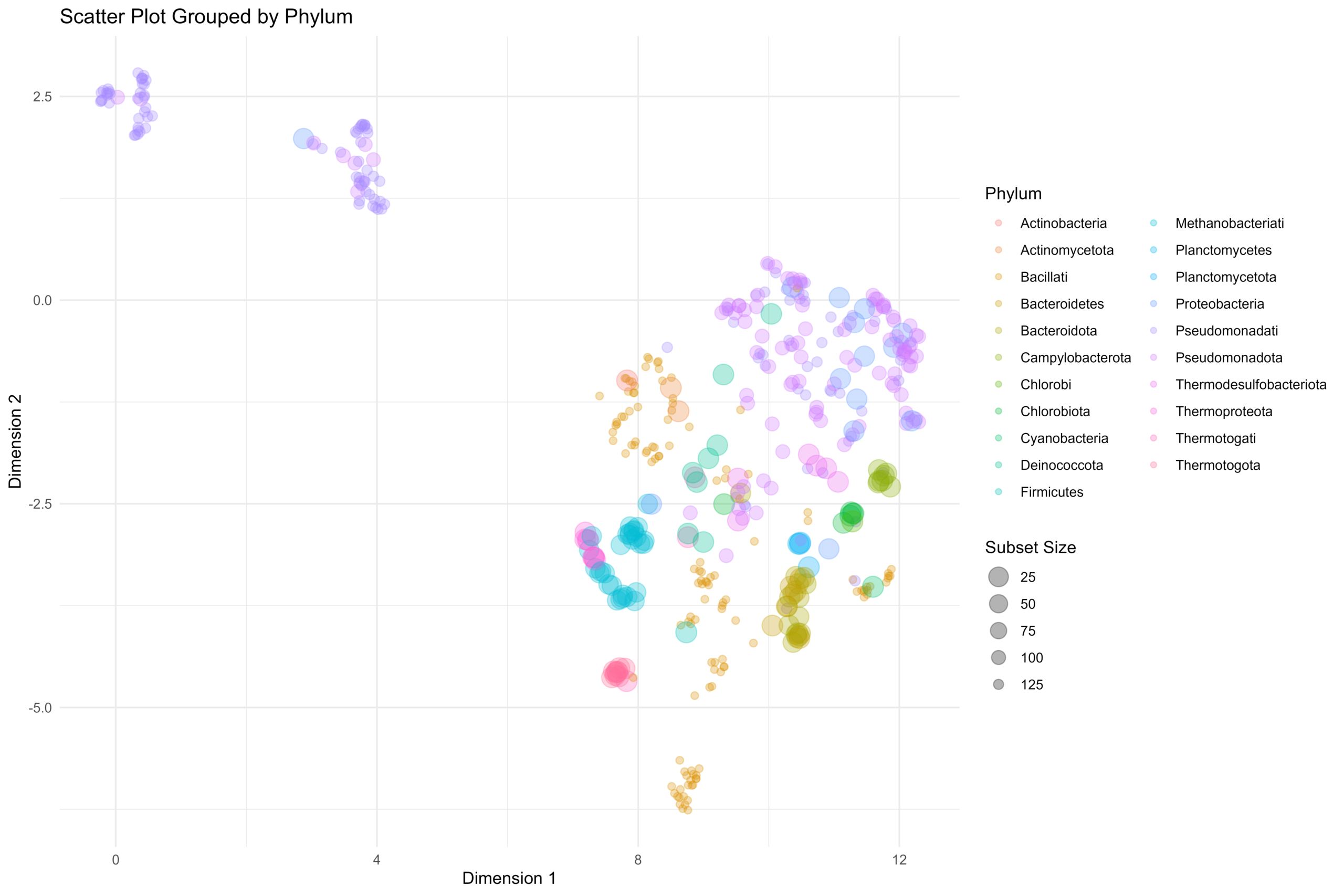
估计阅读时长: 18 分钟在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。除了针对降维后的数据进行散点图可视化,我们还可以直接针对向量化嵌入后的原始嵌入矩阵进行聚类,完成聚类结果的可视化。在这里我们主要是基于嵌入的原始结果进行二叉树聚类可视化。 Order by Date Name Attachments community_metabolic_tree • 220 kB • 392 click 2026年2月15日community-local • […]
估计阅读时长: 17 分钟EC Number是国际酶学委员会(IUBMB)制定的一套酶分类编号体系,EC Number采用层级分类法,由4个数字组成,分别代表酶的大类、亚类、亚亚类和序号。例如,“EC 1.1.1.37”中,第一个“1”表示氧化还原酶大类;第二个“1”表示作用于CH-OH基团;第三个“1”表示以NAD+或NADP+为受体的酶;第四个“37”表示特定酶苹果酸脱氢酶。这种层次结构意味着EC编号蕴含了丰富的功能信息,包括酶催化的反应类型和底物/机制。将EC Number嵌入为向量,有助于我们利用机器学习模型进行功能预测、相似性分析等。 Order by Date Name Attachments Capture • 14 kB • 470 […]
估计阅读时长: 11 分钟在将生物序列(如基因组或蛋白质序列)或文本数据转换为数值向量形式时,TF-IDF(Term Frequency-Inverse Document Frequency)和N-gram One-hot(又称Bag-of-n-grams)是两种经典且基础的文档嵌入算法。它们各自侧重于不同的特征提取方式,常被用于自然语言处理和生物信息学领域。 Attachments scatter_plot • 433 kB • 505 click 2026年2月10日
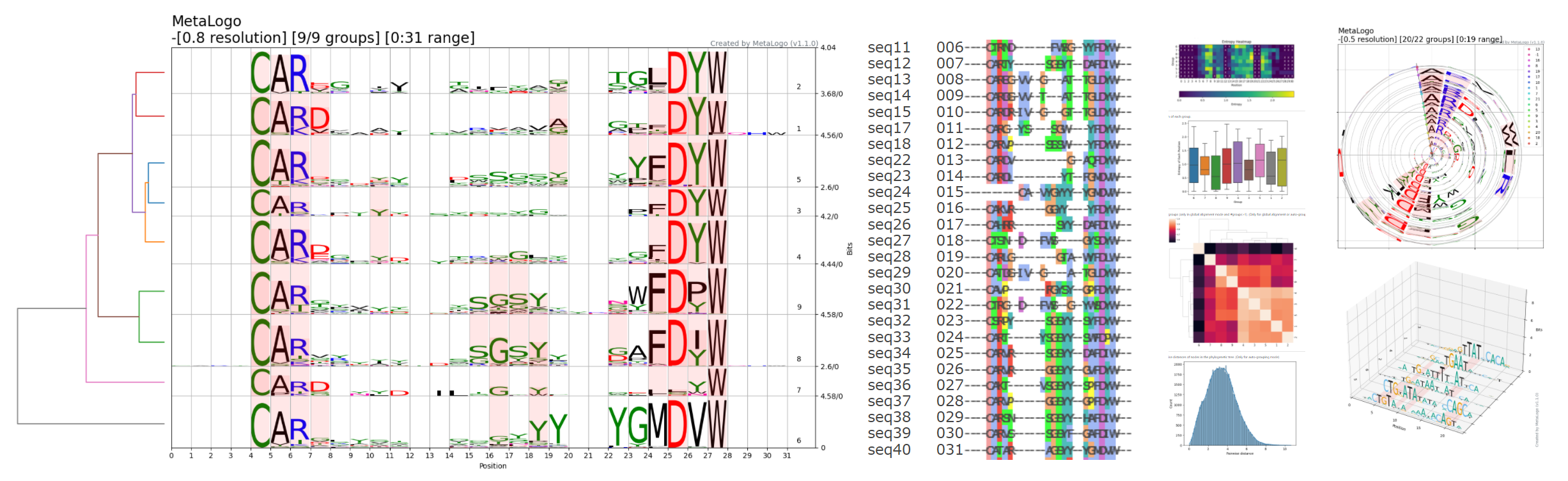
估计阅读时长: 23 分钟Sequence Logo 是一种可视化 DNA 或蛋白质序列保守性的图形表示方法。每个位置(列)上的字母堆叠高度代表该位点的信息含量(以 bits 为单位),而每个字母的高度则与其在该位点出现的频率成正比。高信息量的位置字母堆得高,低信息量的位置则矮甚至接近零。Sequence Logo的绘制遵循信息熵原理,我们可以很直观的通过某一个位置的总高低来了解该处位置的信息含量有多少,高信息量的位置,字母堆的高,一般会出现某一个字符特别高,表明该处非常保守。 位置权重矩阵(Position Weight Matrix, PWM)是描述基因组调控因子结合位点序列模式的核心模型。它通过统计在结合位点序列中每个位置上各核苷酸(或氨基酸)出现的频率,来量化该位置对不同碱基的偏好程度。PWM通常以矩阵形式表示,行对应核苷酸(A、C、G、T/U),列对应序列中的位置,矩阵元素即为该位置该核苷酸相对于背景的权重得分。这一模型简洁且易于计算,因此在转录因子结合位点(TFBS)等调控元件的识别和表征中被广泛采用。 Order by Date Name […]
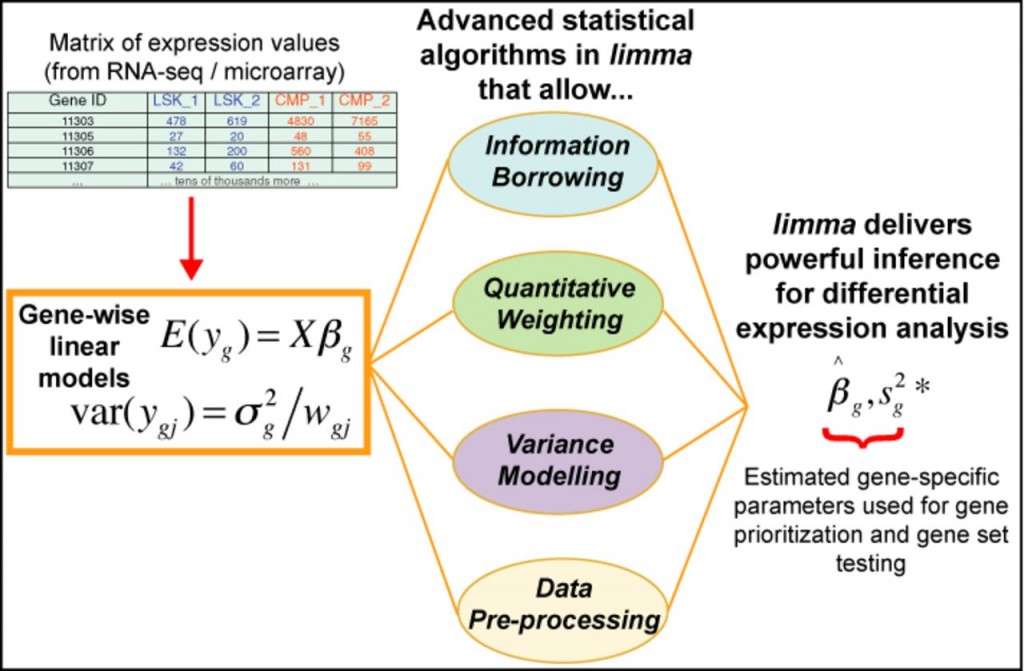
估计阅读时长: 22 分钟limma(Linear Models for Microarray Data)是一个基于R语言的Bioconductor包,最初用于微阵列数据的差异表达分析,现已扩展支持RNA-seq数据。其核心思想是利用线性模型(Linear Models)对基因表达数据进行建模,并结合经验贝叶斯(Empirical Bayes)方法在小样本情况下增强统计推断的稳健性。 Order by Date Name Attachments limma • 119 kB […]

估计阅读时长: 30 分钟零分布(null distribution)是指在假设零假设(null hypothesis)成立的情况下,某个统计量随机取值的概率分布。在统计假设检验中,我们通常提出一个零假设(例如“两组数据没有显著差异”或“观察到的模式仅由随机因素造成”),然后根据观测数据计算一个检验统计量。零分布描述了这个统计量在零假设为真时的分布情况。通过将实际观测到的统计量与零分布进行比较,我们可以计算出P-value:即在零假设下,出现等于或更极端观测结果的概率。如果P-value很小(例如低于预设的显著性水平α),我们就认为零假设不太可能成立,从而拒绝零假设,认为观测结果是统计显著的。 Order by Date Name Attachments image-2 • 66 kB • 570 click 2025年12月16日NULL-pvalue […]

估计阅读时长: 24 分钟假若现在有两条Fasta序列放在你面前,现在需要你进行这两条Fasta序列的相似度计算分析。如果对于我而言,大学刚毕业刚入门生物信息学的时候,可能只能够想到通过blast比对的方式进行序列相似性计算分析。基于blast比对方式可以找到生物学意义上的序列相似性结果,但是计算的效率会比较低。假设现在让你使用这些序列进行机器学习建模分析,或者基于传统数学意义上的基于相似度的无监督聚类分析的时候,面对这些长度上长短不一的生物序列数据,可能会比较蒙圈,因为传统的数学分析方法都要求我们分析的目标至少应该是等长的向量数据。 Order by Date Name Attachments Fasta-A • 544 kB • 937 click 2023年6月29日visualize • 45 […]
估计阅读时长: 5 分钟在工作之中可能会遇到需要进行两个网络图对象之间的相似度计算的情形:例如在质谱数据分析的化学信息学计算工作之中,我们在解析SMILES字符串得到分子图之后,可以基于图相似度比较计算方法来比较计算两个代谢物分子图之间的结构上的相似度。 Attachments pone.0078360.g003 • 2 MB • 1046 click 2022年8月6日https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0078360
估计阅读时长: 4 分钟在代谢组学领域内,LCMS原始数据分析一般分为非靶向全扫原始数据,以及仅针对某些离子进行扫描的MRM靶向质谱数据。虽然二者都是基于LCMS方法进行实验,但是MRM靶向数据由于在事先已经通过实验确定,得到了Q1和Q3离子对信息,所以可以仅针对某一些特定代谢物进行检测。因为MRM数据是针对于某些代谢物检测的靶向数据,所以其XIC谱图在没有同分异构体存在的情况下,一般是很纯净的目标化合物的检测结果数据。所以在原始数据分离,定量计算方面都要比非靶向全扫结果数据要容易很多。 Order by Date Name Attachments xcms-logo-white • 183 kB • 957 click 2022年7月1日lcmspreproc_slides_1.2 • 136 […]

[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 我们在基于前面所论述的《通过diamond软件进行blastp搜索》对大规模的基因组数据进行了代谢酶的EC number的注释以及按照文章《基因组功能注释(EC Number)的向量化嵌入》的方法,得到了一个比较大的基因组代谢酶TF-IDF嵌入丰度矩阵后,如果将这里所得到的嵌入结果矩阵中的基因组,基于Family层级的物种分类分组看作为单细胞转录数据中的细胞分群结果,能否基于单细胞数据分析方法来分析和可视化我的基因组功能嵌入的结果矩阵呢? […]
[…] 对于基于ec number来生成层级数据,我们直接使用《酶EC编号结构解析》文章末尾所展示的层级数据生成函数来实现。 […]
[…] 在前面的一篇《基因组功能注释(EC Number)的向量化嵌入》博客文章中,针对所注释得到的微生物基因组代谢信息,进行基于TF-IDF的向量化嵌入之后。为了可视化向量化嵌入的效果,通过UMAP进行降维,然后基于降维的结果进行散点图可视化。通过散点图可视化可以发现向量化的嵌入结果可以比较好的将不同物种分类来源的微生物基因组区分开来。 […]
😲啊?