
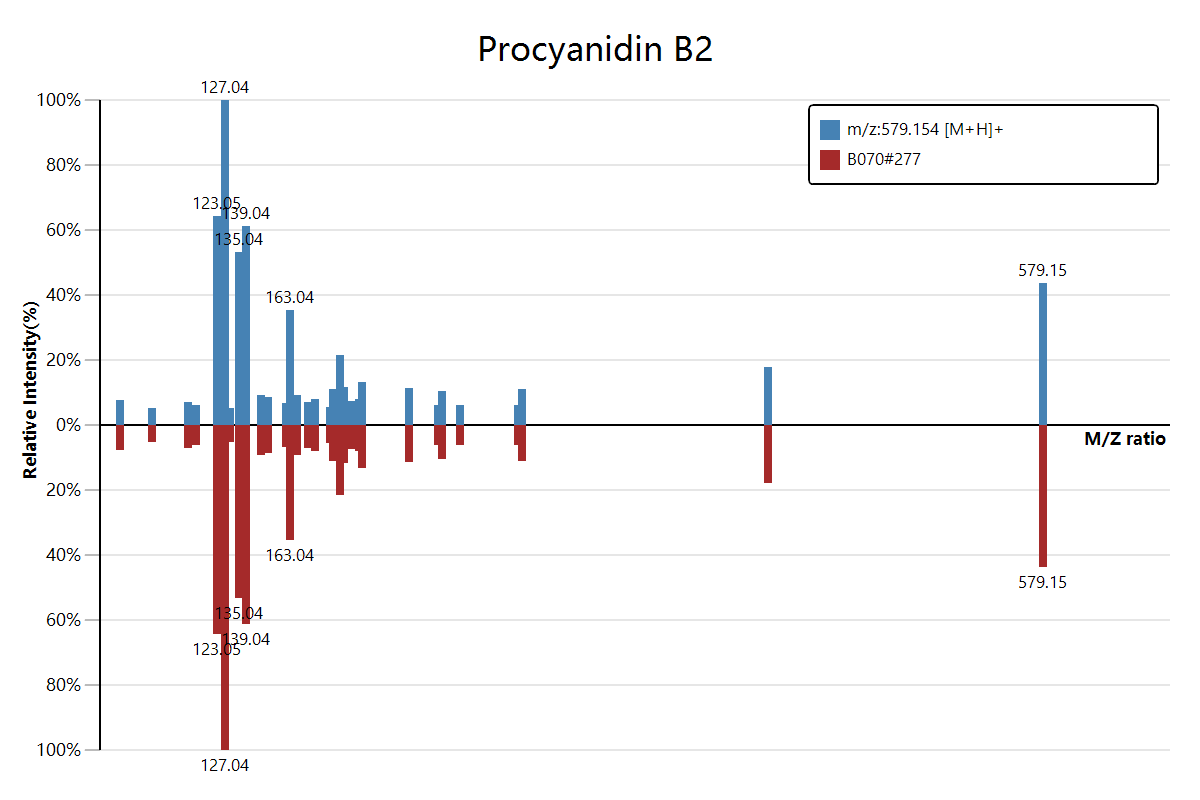
估计阅读时长: 2 分钟https://mzkit.org/ 代谢组文章整个坎坷的发表经历中,大家可能都会遇到的一个老大难问题就是我们有时候会需要从原始数据中得到物质注释结果的二级质谱图数据。对于熟悉xcms程序包的同学,获取二级质谱图可能会比较容易:无非就是加载原始数据,然后按照m/z和rt找到对应的二级scan就好了。但是,这种方法会需要编写脚本来完成。 Order by Date Name Attachments download • 33 kB • 524 click 2021年8月4日[278][MS_MS] FTMS […]

估计阅读时长: 2 分钟https://github.com/dotvanilla/vanilla 在Vanilla编译器项目之中,会需要一个程序模块将VisualBasic代码进行解析为语法树。然后我们基于此语法树就可以将VisualBasic项目转换为WAST源代码,从而实现编译为WebAssembly程序了。在这个步骤之中,我们可以通过一个微软官方的Roslyn编译器平台来实现。 Order by Date Name Attachments Roslyn-nuget • 107 kB • 498 click 2021年7月24日what-is-visual-studio • […]

估计阅读时长: < 1 分钟https://github.com/xieguigang/codegraph Attachments Microsoft.VisualBasic.Framework_v47_dotnet_8da45dcd8060cc9a.dll • 10 MB • 429 click 2021年8月29日
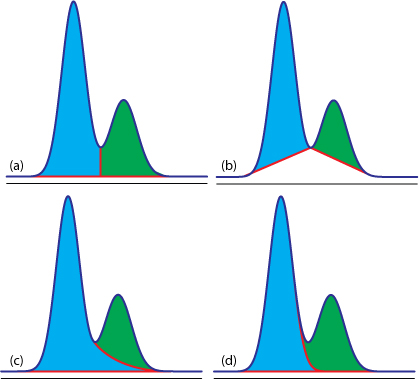
估计阅读时长: 8 分钟https://github.com/xieguigang/sciBASIC/tree/master/Data_science/Mathematica/SignalProcessing 进行峰识别是在代谢组学原始数据分析之中进行定量分析的很重要的一环。在代谢组学之中,定量分析分为靶向定量,以及非靶向定量计算这两大部分。 Order by Date Name Attachments Figure12.36 • 50 kB • 541 click 2021年7月10日view_signal • […]

估计阅读时长: 9 分钟https://github.com/dotvanilla/vanilla WebAssembly是一种运行在浏览器端的二进制程序集文件。和普通的应用程序开发一样,WebAssembly需要基于一定的源代码文本进行编译。这个编译所需要的源代码文本就是WAST文件。 Understanding WebAssembly text format(https://developer.mozilla.org/en-US/docs/WebAssembly/Understanding_the_text_format)
估计阅读时长: 4 分钟https://github.com/dotvanilla/vanilla vanilla编译器项目是我之前开发过的一个实验性质的项目。主要是为了解决在浏览器端的一些高性能计算的需求,例如数据加密和解密,基于WebGL的计算机图形项目,力学物理规律模拟,网络可视化布局计算等。 Order by Date Name Attachments 1_PcKt44c-UZBBTfNBaovxeQ • 49 kB • 463 click 2021年7月8日web-assembly-architecture-xenonstack-3-1 • […]
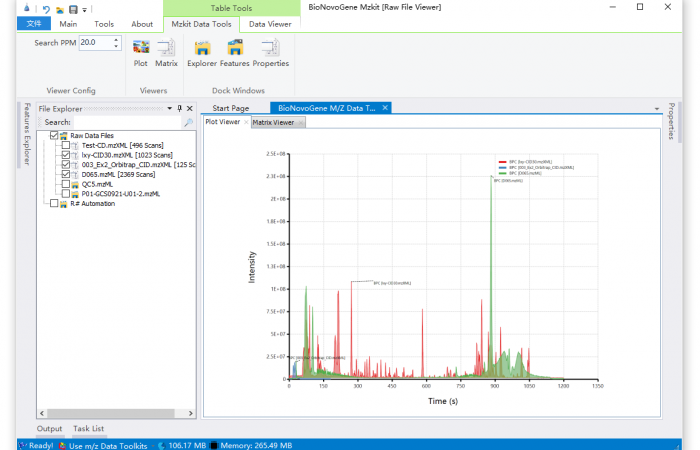
估计阅读时长: 3 分钟https://mzkit.org mzkit软件是我最近开发的一款开源的代谢组学领域内的原始数据文件查看工具。开发mzkit软件的初衷是为了更方便的查看很大的非靶向原始数据文件:因为在开发mzkit软件之前,在开发LCMS的代谢物注释脚本或者建立标准品库数据的时候,如果我想要查看或者导出文件中的一些质谱图碎片信息,会需要通过R环境之中的xcms程序包编程来完成。通过R脚本来查看原始数据文件,非常的不方便。所以就有了mzkit软件项目的诞生。 Order by Date Name Attachments BPC_overlay • 114 kB • 581 click 2021年7月1日LCMS_scanTree • […]
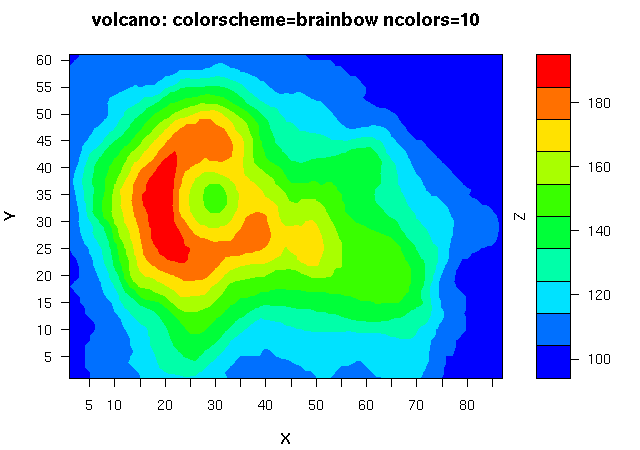
估计阅读时长: 16 分钟https://github.com/xieguigang/sciBASIC 等高线指的是地形图上高程相等的相邻各点所连成的闭合曲线。把地面上海拔高度相同的点连成的闭合曲线,并垂直投影到一个水平面上,并按比例缩绘在图纸上,就得到等高线。 Order by Date Name Attachments 1_Contour • 487 kB • 653 click 2021年6月30日Ms1Contour • […]
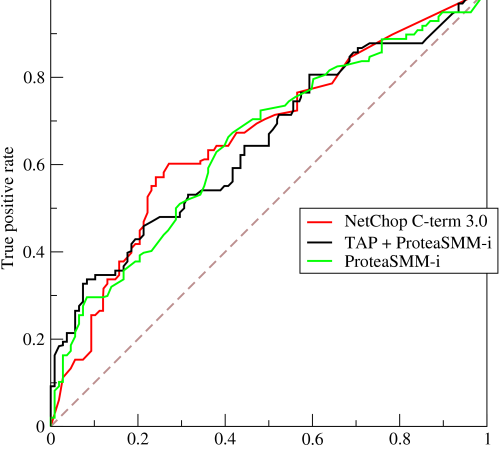
估计阅读时长: 8 分钟https://github.com/rsharp-lang/R-sharp 对于0,1两类分类问题,一些分类器得到的结果往往不是0,1这样的标签。如神经网络得到诸如0.5,0.8这样的分类结果。这时,我们人为取一个阈值,比如0.4,那么小于0.4的归为0类,大于等于0.4的归为1类,可以得到一个分类结果。同样,这个阈值我们可以取0.1或0.2等等。 Order by Date Name Attachments ROC • 221 kB • 589 click 2021年6月28日Roccurves • […]
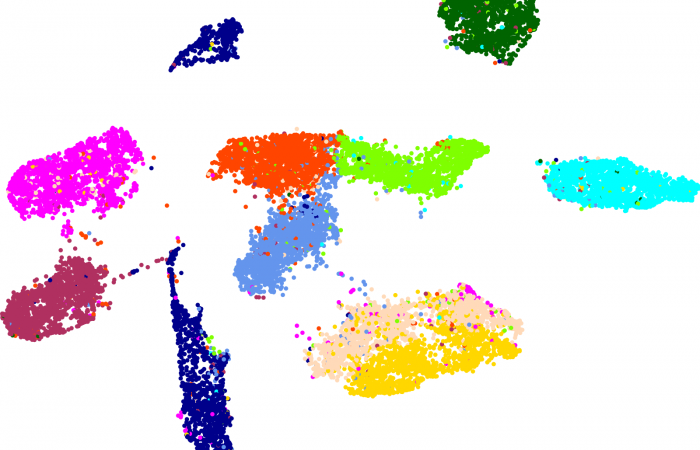
估计阅读时长: 23 分钟https://github.com/rsharp-lang/R-sharp 降维是将数据由高维约减到低维的过程而用来揭示数据的本质低维结构。它作为克服“维数灾难”的途径在这些相关领域中扮演着重要的角色。在过去的几十年里,有大量的降维方法被不断地提出并被深入研究,其中常用的包括传统的降维算法如PCA和MDS;流形学习算法如UMAP、t-SNE、ISOMAP、LE以及LTSA等。 Order by Date Name Attachments MNIST-LabelledVectorArray-60000x100 • 230 kB • 681 click 2021年6月27日MNIST-LabelledVectorArray-60000x100Euclidean_Distance • […]

Hello blogger, thank you for sharing this post! We process a large number of metagenomic samples, and every time we…
谢博,您好。阅读了您的博客文章非常受启发!这个基于k-mer数据库的过滤框架,其核心是一个“污染源数据库”和一个“基于覆盖度的决策引擎”。这意味着它的应用远不止于去除宿主reads。 我们可以轻松地将它扩展到其他场景: 例如去除PhiX测序对照:建一个PhiX的k-mer库,可以快速剔除Illumina测序中常见的对照序列。 例如去除常见实验室污染物:比如大肠杆菌、酵母等,建一个联合的污染物k-mer库,可以有效提升样本的纯净度。 例如还可以靶向序列富集:反过来想,如果我们建立一个目标物种(比如某种病原体)的k-mer库,然后用这个算法去“保留”而不是“去除”匹配的reads,这不就实现了一个超快速的靶向序列富集工具吗? 这中基于kmer算法的通用性和扩展性可能会是它的亮点之一。感谢博主提供了这样一个优秀的思想原型
It’s laborious to find knowledgeable people on this topic, however you sound like you realize what you’re speaking about! Thanks
WOW, display an image on a char only console this is really cool, I like this post because so much…
确实少有, 这么高质量的内容。谢谢作者。;-) 我很乐意阅读 你的这个技术博客网站。关于旅行者上的金唱片对外星朋友的美好愿望,和那个时代科技条件限制下人们做出的努力,激励人心。